
Seltene Erkrankungen
Seltene Erkrankungen sind nicht so publikumswirksam wie Bluthochdruck, Diabetes oder Depressionen. Sie betreffen jeweils nur wenige Menschen in Deutschland und Europa und im Extremfall gibt es nur wenige Familien weltweit.
Diese Erkrankungen werden deshalb auch Orphan Diseases genannt. Orphan bedeutet im englischen „Waise“ oder „Waisenkind“. Tatsächlich sind häufig Kinder betroffen. „Leider sind die Seltenen Erkrankungen noch immer in weiten Teilen die Stiefkinder der Medizin“, sagt Prof. Annette Grüters-Kieslich, Vorstandsvorsitzende der Eva-Luise- und Horst-Köhler-Stiftung, die sich für Menschen mit Seltenen Erkrankungen einsetzt.
Die Erkrankungen sind oft so selten, dass Ärztinnen und Ärzte sie nicht kennen und deshalb auch nicht erkennen können: Der Weg zur Diagnose dauert oft viele Jahre, begleitet von Fehldiagnosen und falschen Therapien. Ist die Diagnose endlich gestellt, bedeutet das noch lange nicht, dass die Krankheit behandelbar ist. Nur für zwei Prozent der Orphan Diseases sind Arzneimittel verfügbar – sogenannte Orphan Drugs. Es braucht also noch viel Forschung für neue Medikamente, und diese braucht Förderung. Denn die pharmazeutischen Unternehmen gehen ein großes Risiko ein, wenn sie sich der Forschung und Entwicklung von Orphan Drugs widmen, weil es nur so wenige Betroffene gibt. Doch es lohnt sich, für jede einzelne Patientin, für jeden einzelnen Patienten.
Wie immer können Sie das Text- und Bildmaterial des Themendienstes gerne unter Nennung der Quellen kostenlos redaktionell verwenden.
Ihr BPI-Presseteam
Seltene Erkrankungen sind zusammen genommen viele, nämlich mehr als 8.000 ganz verschiedene Krankheiten. „Durch die rasante Entwicklung neuer Methoden und Verfahren in der Medizin entdecken Forscherinnen und Forscher momentan quasi im Wochentakt neue, zumeist genetisch bedingte, seltene Krankheiten“, berichtet Prof. Annette Grüters-Kieslich, Vorstandsvorsitzende der Eva-Luise- und Horst-Köhler- Stiftung für Menschen mit Seltenen Erkrankungen in Bonn.
Von selten bis extrem selten
Manche Schätzungen gehen von etwa 10.000 Seltenen aus. Insgesamt leiden in Deutschland immerhin etwa vier Millionen Menschen an einer solchen Erkrankung, das sind etwa sechs bis sieben Prozent der Bevölkerung. Wie selten ist selten? Dafür gibt es eine Definition in Zahlen: Als selten gilt eine Erkrankung in der Europäischen Union gemäß der Verordnung (EG) 141/2000, wenn nicht mehr als fünf von 10.000 Menschen betroffen sind. Zum Vergleich: Von 10.000 Menschen leiden 750 an Diabetes Typ 2.
Dabei ist die Spannbreite innerhalb der Seltenen Erkrankungen groß: Manche sind recht bekannt und gehören zu den häufiger vorkommenden Seltenen Erkrankungen, wie zum Beispiel die Mukoviszidose – eine angeborene Stoffwechselerkrankung – oder Eierstockkrebs. Bei beiden Erkrankungen gibt es etwa ein bis fünf Betroffene auf 10.000 Menschen. Extrem selten und unbekannt ist dagegen zum Beispiel das Hutchinson-Gilford-Syndrom – eine angeborene Erkrankung, bei der aufgrund eines Gendefekts schon in frühester Kindheit Haut, Skelett und Blutgefäße vorzeitig altern. Statistisch gesehen sind nur 0,005 pro 100.000 Menschen davon betroffen. Zudem gibt es Tausende von Seltenen Erkrankungen, bei denen nur wenige Fälle weltweit bekannt sind. Die Gaucher-Krankheit, an der Sabine Biermann erkrankt ist (siehe Fallbeispiel), kommt bei 1,7 pro 100.000 Menschen vor.
Alle Organe können betroffen sein
Auch die Spannbreite der Symptome und Fachgebiete ist groß, weil alle Organe betroffen sein können. Ärztinnen und Ärzte aller Fachrichtungen sind mit Seltenen Erkrankungen konfrontiert, wie zum Beispiel aus der Allgemeinmedizin, Inneren Medizin, Kardiologie, Rheumatologie, Pulmologie, Onkologie, Infektiologie, Kinder- sowie Augenheilkunde.
Die meisten Seltenen Erkrankungen, nämlich rund 80 Prozent, beruhen auf erblichen Gendefekten. So ist zum Beispiel bei der spinalen Muskelatrophie das Gen SMN1 beschädigt, so dass die Kinder Muskelschwund entwickeln. Beim Hutchinson-Gilford-Syndrom verursacht das defekte LMNA-Gen den vorzeitigen Alterungsprozess. Bei der Leberschen Kongenitalen Amaurose, die zur Blindheit führen kann, sind es Mutationen im RPE65-Gen, die zu einer Degeneration der Netzhaut führen.
Zudem gehören bestimmte Krebsarten zu den Seltenen, so etwa Knochenmarkskrebs (Multiples Myelom), Krebs des Lymphsystems, im Gallengang, an der Niere oder eben in den Eierstöcken. Auch Infektionskrankheiten wie die Zytomegalovirus-Infektion, Autoimmunkrankheiten wie die Riesenzell-Arteriitis oder Herz-Kreislauf-Erkrankungen wie der Lungenhochdruck sind in der langen Liste der Seltenen Erkrankungen zu finden.
Die meisten Seltenen Erkrankungen brechen im Kindesalter aus. „Viele betroffene Kinder werden niemals ein selbstständiges Leben führen können, mindestens 1.000 versterben jährlich allein in Deutschland“, sagt die Kinderärztin Prof. Grüters-Kieslich. Denn nur für 160 der 8.000 seltenen Erkrankungen gibt es mittlerweile ursächliche Therapien und spezifische Medikamente. „Leider sind die Seltenen Erkrankungen noch immer in weiten Teilen die Stiefkinder der Medizin“, so Grüters-Kieslich. „Es fehlt an strukturierten Versorgungswegen, an Spezialistinnen und Spezialisten, an Investitionen in die Forschungsstruktur. So dauert es meist viele Jahre bis zur Diagnose und oft kann nicht gezielt behandelt werden.“
„Wenn ich morgens aufgestanden bin, geduscht und gefrühstückt habe, musste ich mich schon wieder hinlegen“, berichtet die 58-jährige Sabine Biermann. Im Jahr 2004 fiel dann endlich die Diagnose: Morbus Gaucher. Doch bereits 1991 wies ihre Frauenärztin sie darauf hin, dass die Anzahl der Blutplättchen im Blut bei ihr extrem niedrig ausfällt. Es bestand damals der Verdacht auf Blutkrebs, deshalb wurde die Berlinerin in eine Universitätsklinik überwiesen. Dabei stellte sich heraus, dass ihre Milz und Leber vergrößert waren. Sie litt an deutlichen Symptomen: Sie war immer sehr müde und ihre Knochen schmerzten.
Falscher Verdacht
Morbus Gaucher ist eine seltene Stoffwechselerkrankung, bei der sich durch einen Enzymmangel spezielle Moleküle des Fettstoffwechsels in Leber, Milz, Knochenmark und anderen Organen anreichern. Das ist typisch für Seltene Erkrankungen: Es sind oft viele Organe betroffen, entsprechend vielfältig sind die Symptome, was Ärztinnen und Ärzte oft Rätsel aufgibt. Typisch ist auch, dass Sabine Biermann die Erkrankung von ihren Eltern „geerbt“ hat: Sowohl ihre Mutter als auch ihr Vater hatten dieselbe Genmutation. Damals war Morbus Gaucher noch nicht so bekannt wie heute. Immer wieder wurde Sabine Biermann in der Berliner Charité untersucht. Es wurden Computertomografien und Ultraschalluntersuchungen angewiesen, sowie das Knochenmark punktiert. Nur dank eines umsichtigen Assistenzarztes blieb ihr eine Chemotherapie erspart.
Alles nur psychisch?
Im Jahr 1996 – seit dem ersten auffälligen Befund sind inzwischen fünf Jahre vergangen – nahm ein Onkologe einer anderen Klinik zum ersten Mal die Verdachtsdiagnose „Morbus Gaucher“ in den Mund. Doch die Unsicherheit der niedergelassenen Ärztinnen und Ärzte, die Sabine Biermann daraufhin konsultierte, war groß. Eine Hausärztin lehnte es ab, sie mit einem Medikament gegen Morbus Gaucher zu behandeln, das es damals schon gab. Weil die Diagnose nicht sicher und das Arzneimittel so teuer war. Ein anderer Arzt schob alles auf die Psyche. Inzwischen verstärkten sich die Symptome: Zu Müdigkeit und Knochenschmerzen kamen Magen-Darm-Beschwerden hinzu. Frau Biermann stieg aus dem Beruf aus, und kümmerte sich nur noch um die beiden Töchter.
„Wundermittel“ Enzymersatztherapie
Schließlich stieß ihr Mann im Internet auf die Gaucher-Gesellschaft Deutschland (GGD) e.V., die ihr Spezialisten empfahl. Mit Hilfe einer genetischen Diagnostik bestätigte sich schließlich im Jahr 2004 der Verdacht von Morbus Gaucher; der Experte empfahl eine Enzymersatztherapie (s. Kapitel IV). Frau Biermann fand einen Arzt in Berlin, der ihr die Infusionen alle zwei Wochen verabreichte.
„Das war wie ein Wundermittel, es ging richtig bergauf“, berichtet Biermann, die heute kaum noch Beschwerden hat. Ab und zu muss sie wegen Erschöpfung mal einen Nachmittag kürzertreten, und ab und zu spürt sie ihre Knochen. Sie arbeitet als Praxismanagerin in einer großen Augenarztpraxis, sie geht zum Yoga, zu Pilates und zum Gerätetraining – und fungiert ehrenamtlich als stellvertretende Vorstandsvorsitzende bei der GGD.
Ärztinnen und Ärzte bekommen es mit aller Wahrscheinlichkeit nur zwei- bis dreimal in ihrem Berufsleben mit einer Seltenen Erkrankung zu tun. „Die Erkrankungen kommen so selten vor, gleichzeitig sind es so viele – keine Ärztin, kein Arzt kann sich 6.000 oder 8.000 Erkrankungen merken“, sagt Prof. Annette Grüters-Kieslich von der Eva-Luise- und Horst-Köhler-Stiftung für Menschen mit Seltenen Erkrankungen. Zudem fehle es bei den meisten dieser Leiden an klaren Leitsymptomen. „Häufig tauchen Symptome auf, die eigentlich gar nicht zusammenpassen.“
Bei Sabine Biermann, die an der Gaucher-Erkrankung leidet (siehe Fallbeispiel), waren es einerseits die vergrößerte Milz und Leber, andererseits das veränderte Blutbild – Symptome, die auf den ersten Blick nichts miteinander zu tun haben. „Die Patientinnen und Patienten müssen viele Hürden überwinden, bis sie an den richtigen Spezialisten, die richtige Spezialistin gelangen“, sagt Biermann, die Patientin und gleichzeitig stellvertretende Vorsitzende bei der Gaucher-Gesellschaft Deutschland ist. Fehl- und Doppelbehandlungen sind die Folge, „mitunter werden kritische Zeitfenster für eine rechtzeitige Intervention verpasst, mit dann natürlich besonders tragischen Folgen für die Betroffenen“, ergänzt Grüters-Kieslich.
Das Projekt Translate-NAMSE
Um den Weg zur richtigen Diagnose zu verkürzen, haben sich sogenannte Zentren für Seltene Erkrankungen gegründet. Davon gibt es inzwischen bundesweit 37; einen Überblick bietet die Seite se-atlas.de. Dass sich solche übergeordneten, interdisziplinär arbeitenden Zentren etablieren, ist ein Hauptanliegen des Nationalen Aktionsbündnisses für Menschen mit Seltenen Erkrankungen (NAMSE), zu dem auch der BPI gehört (s. Interview). Um das Zentrenmodell voranzutreiben, haben sich in dem Projekt Translate-NAMSE neun Zentren an Universitätskliniken und vier universitäre humangenetische Institute miteinander vernetzt.
Interdisziplinäre Fallkonferenzen
Der Gemeinsame Bundesausschuss (G-BA) hat das dreijährige Projekt, das vom Innovationsfonds des G-BA gefördert wurde, im April 2022 als erfolgreich bewertet. Denn von den etwa 6.000 rekrutierten Patienten konnte immerhin bei einem Drittel eine gesicherte Diagnose gestellt werden. „Das ist ein großartiger Erfolg“, berichtet Prof. Annette Grüters-Kieslich. „Insbesondere die interdisziplinären Fallkonferenzen haben sich als sehr hilfreich erwiesen, um bei komplexen Fällen weiterzukommen.“ Die Kinderärztin war federführend an der Entwicklung des Projekts Translate-NAMSE beteiligt.
Neben Kinderärzten sitzen in den Fallkonferenzen auch Expertinnen und Experten der Humangenetik, Neurologie, Onkologie, Inneren Medizin und weitere. „Viele Patienten, die zuvor jahrelang fehlbehandelt durch das Gesundheitswesen irrten, haben im Rahmen des Projekts innerhalb eines halben Jahres eine präzise Diagnose erhalten“, so Grüters-Kieslich. Die Ärzte-Odyssee endet bei den Kindern im Schnitt nach viereinhalb Jahren, bei den Erwachsenen nach achteinhalb Jahren.
Innovative Diagnostik
Insbesondere die sogenannte Exomsequenzierung hat sich als praktikable Diagnosemethode erwiesen, deshalb übernehmen mehrere Krankenkassen bereits die Kosten im Rahmen von Selektivverträgen.
Bei der Exomsequenzierung werden die ein bis zwei Prozent des menschlichen Genoms untersucht, in denen 85 Prozent der bekannten krankheitsverursachenden Varianten zu finden sind. Am Dr.-von- Haunerschen-Kinderspital an der Universitätsklinik München – ein Zentrum für Seltene Erkrankungen – sequenzieren die Wissenschaftler schon seit über zehn Jahren die Gene ihrer kleinen Patienten.
„Wir haben damals ein riesiges Ungetüm gekauft, dessen Genauigkeit noch nicht überzeugend war“, erzählt der Ärztliche Direktor Prof. Christoph Klein. Inzwischen ist man bei der Sequenzierung der dritten Generation angelangt, dem sogenannten Next Generation Sequencing, kurz NGS (s. auch Interview). „Mit Hilfe des NGS lassen sich die Gene wesentlich schneller und kostengünstiger sequenzieren. Zudem zeigen die Geräte höchste Präzision im Ablesen von genetischer Information“, berichtet Prof. Klein. Bringt die Exomsequenzierung keine befriedigenden Ergebnisse, kommt auch eine Sequenzierung des gesamten Genoms zum Zug. „Wir wollen bei mehr Kindern eine klare Diagnose erreichen“, so Klein. Auch wenn dann kein gezieltes Orphan-Drug-Medikament (s. Kapitel Orphan Drugs) zur Verfügung steht, kann nach Identifizierung des ursächlichen Gens die Therapie besser auf die Symptome abgestellt werden.
Mangelnde Finanzierung
Prof. Klein weist darauf hin, dass er die Diagnose-Geräte mit Hilfe von Spenden für die Stiftung „Care for Rare Foundation“ finanziert hat. Der Kinderarzt und Spezialist für Pädiatrische Immunologie, Hämatologie und Onkologie hat die Stiftung gegründet, die sich der internationalen Vernetzung verschrieben hat (https://www.care-for-rare.org). „Dass es die Zentren für Seltene Erkrankungen gibt, heißt ja noch lange nicht, dass sie auch finanziert sind“, so Klein. „Wir kämpfen seit 15 Jahren um Zuschläge.“ Die Kinder bräuchten nicht nur einen, sondern eine ganze Reihe von Spezialistinnen und Spezialisten. „Das sind Vorhaltekosten, die im normalen Krankenversicherungssystem nicht abgebildet sind.“ (s. Interview). Er bemängelt zudem, dass zum Beispiel eine umfassende psychosoziale Betreuung für die schwer kranken Kinder in den Kliniken nicht vorgesehen ist. Das sei „ein Riesen-Problem“.
Neugeborenen-Screening
Eine besonders frühe Diagnostik stellen die Screenings von Neugeborenen dar. „Die Screenings erlauben uns, schwere Störungen frühzeitig zu entdecken und betroffene Kinder umgehend behandeln zu können“, sagt Prof. Annette Grüters-Kieslich. „In vielen Fällen ist das entscheidend dafür, dass die Kinder überleben und sich normal entwickeln.“ Inzwischen wird für 17 Krankheiten ein solches Screening angeboten, darunter angeborene Stoffwechselerkrankungen und Hormonstörungen. „Grundsätzlich gilt, dass ein Neugeborenen-Screening nur dann Sinn macht, wenn sich daraus auch eine Behandlungsoption ergibt, die die Prognose des Kindes entscheidend verbessert“, so Grüters-Kieslich. Dank vieler neuer Therapieansätze kommen immer mehr Krankheiten für ein Screening in Frage: So ist unlängst auch ein Screening für die Sichelzellkrankheit und die spinale Muskelatrophie eingeführt worden.
Nur zwei Prozent der Seltenen Erkrankungen sind ursächlich behandelbar. Das heißt, für 98 Prozent gibt es bislang noch keine Therapie. „Da die allermeisten Seltenen Erkrankungen nahezu unerforscht sind, bleiben Therapiemöglichkeiten in der Regel auf die Behandlung von Symptomen beschränkt“, sagt Prof. Annette Grüters-Kieslich von der Eva-Luise- und Horst-Köhler-Stiftung für Menschen mit Seltenen Erkrankungen. „Kurative Behandlungsansätze sind ebenso wie spezifische Arzneimittel weiterhin die Ausnahme.“
Doch solche Ausnahmen gibt es und die werden mehr. Rund ein Drittel der Arzneimittel, die in den vergangenen fünf Jahren neu auf den Markt kamen, sind Medikamente gegen Seltene Erkrankungen. Derzeit sind, laut Zahlen des BPI, 133 Arzneimittel mit einem Orphan-Drug-Status in der EU zugelassen und stehen für die Behandlung von Seltenen Erkrankungen zur Verfügung.
Hinzu kommen 66 weitere Arzneimittel, die früher einmal einen Orphan-Drug-Status besaßen oder deren Status zehn Jahre nach ihrer Marktzulassung abgelaufen ist. So stehen insgesamt knapp 200 Wirkstoffe zur Behandlung von rund 150 seltenen Leiden zur Verfügung. „Insbesondere bei den Gentherapien zeigen sich erfolgversprechende Ansätze“, berichtet Dr. Matthias Wilken, Geschäftsführer Market Access, Märkte und Versorgung beim BPI. „Ansonsten handelt es sich bei den Orphan Drugs zum Beispiel um Enzymersatztherapien, Hormone, immunstimulierende oder immununterdrückende Substanzen sowie Krebsmedikamente.“
Beispiele für erfolgreiche Therapieansätze mit Orphan Drugs
Ein medizinischer Wirkstoff ist noch kein Arzneimittel und auch ein Wirkstoff fällt nicht fertig vom Himmel, sondern wird aus verschiedenen Grundstoffen gewonnen und durch chemische Reaktionen optimiert.
- Mukoviszidose: eine Erkrankung, die von einem mutierten CFTR-Gen ausgelöst wird. Zäher Schleim verstopft eine Reihe lebenswichtiger Organe. Lag die durchschnittliche Lebenserwartung der Patientinnen und Patienten in den 1980er Jahren noch zwischen 25 und 30 Jahre, ist sie bis zum Jahr 2019 auf über 50 Jahre gestiegen – dank der Entwicklung von CFTR-Modulatoren, die die Funktion des defekten CFTR-Proteins verbessern. Seit 2016 gehört Mukoviszidose auch zum Neugeborenen-Screening, so dass schon bei Säuglingen sofort mit der notwendigen Therapie begonnen werden kann.
- Morbus Gaucher: An dieser seltenen Stoffwechselerkrankung, bei der aufgrund eines Gendefekts ein spezielles Enzym fehlt, leidet Sabine Biermann (siehe Fallbeispiel). Inzwischen stehen zwei verschiedene Therapieansätze zur Verfügung: Zum einen die Enzymersatztherapie, bei der das fehlende Enzym beta-Glukozerebrosidase gespritzt wird. Zum anderen die Substratreduktionstherapie, bei der die Bildung des Stoffwechselprodukts Glukozerebrosid gehemmt wird. Glukozerebrosid sammelt sich im Körper an, weil es nicht von dem fehlenden Enzym beta-Glukozerebrosidase gespalten wird. Es lagert sich in Milz, Leber, Nieren, Lungen, Gehirn und Knochenmark ein, was zu einer Vielzahl von Symptomen führen kann: unter anderem zu einer vergrößerten Milz und Leber, Funktionsstörungen der Leber, Knochenkrankheiten, neurologischen Komplikationen, aufgeblähten Bauch, niedrige Blutplättchenzahl.
- Lebersche Hereditäre Optikus-Neuropathie (LHON): Nervenzellen der Netzhaut (Ganglienzellen) gehen zugrunde. Die Erkrankung raubt die Sehkraft bis hin zur vollständigen Erblindung. Eine frühe Therapie mit einem Orphan Drug, das die Ganglienzellen in der Netzhaut reaktivieren kann, kann die Sehschärfe stabilisieren und das Sehvermögen verbessern.
- Spinale Muskelatrophie: Die spinale Muskelatrophie ist die häufigste genetisch bedingte Todesursache bei Kleinkindern. Weil das Gen SMN1 defekt ist, arbeiten die Nervenzellen im Rückenmark nicht richtig, so dass die Kinder Muskelschwund entwickeln und daran sterben. Mittlerweile stehen Orphan Drugs zur Verfügung mit der Folge, dass die betroffenen Kinder wieder den Kopf heben, selbstständig sitzen, essen und atmen können (s. auch BPI-Themendienst Arzneimittelinnovationen).
Eine Gentherapie kann gerade bei Seltenen Erkrankungen der Schlüssel zu einer ursächlichen Therapie sein, da Seltene Erkrankungen sehr oft durch nur einen Gendefekt verursacht werden. Neben der Spinalen Muskelatrophie lässt sich inzwischen auch ein schwerer Immundefekt bei Kindern gentherapeutisch behandeln, bei dem sich aufgrund eines fehlenden Enzyms keine weißen Blutkörperchen bilden. Auch für die vererbte Augenkrankheit Lebersche Amaurose, die zur Erblindung führt, sowie die seltene Blutererkrankung Beta-Thalassämie gibt es inzwischen ein Gentherapeutikum. Seit Ende 2020 steht auch gegen die Metachromatische Leukodystrophie ein gentherapeutisches Arzneimittel zur Verfügung. Unbehandelt zerstört die Krankheit die schützende Myelinschicht der Nerven, so dass motorische und mentale Fähigkeiten fortschreitend verloren gehen.
Sonderstatus bei der Zulassung
Orphan Drugs sind besondere Medikamente, denn sie haben zunächst mal ein Problem: Da die Entwicklung eines Arzneimittels bis zu 15 Jahre dauern kann und die Investitionen sich auf mehrere Hundert Millionen Euro belaufen, lohnt sich deren Entwicklung angesichts der sehr wenigen Patienten unter normalen Bedingungen für die pharmazeutische Industrie nicht. Deshalb wurden mit der europäischen Orphan-Drug-Verordnung (EG) 141/2000 im Jahr 2000 folgende Anreize für die pharmazeutischen Unternehmen geschaffen:
- Das neue Medikament ist zehn Jahre lang nicht nur vor Nachahmerpräparaten mit dem gleichen Wirkstoff geschützt, sondern auch davor, dass Medikamente mit anderen Wirkstoffen gegen die gleiche Krankheit zugelassen werden – sofern diese keine weiteren Vorteile für die Therapie bringen. Es gibt also ein Jahrzehnt lang eine Marktexklusivität.
- Die Gebühren bei der Zulassung durch die Europäische Arzneimittelagentur EMA werden reduziert, für kleine und mittlere Unternehmen entfallen sie vollständig.
- Zudem erhalten die Unternehmen administrative Unterstützung durch die EMA.
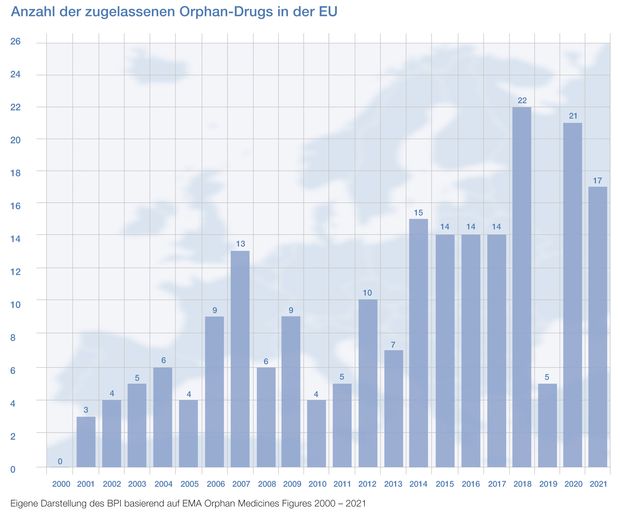
Dass die europäische Verordnung (EG) 141/2000 wirkt, beweisen die Zahlen: Kam in den Jahren 1998 bis 2000 gerade mal ein Orphan Drug pro Jahr auf den Markt, waren es im Jahr 2006 schon neun. In den Jahren 2020 und 2021 wurden zusammen 35 neue Orphan Drugs zugelassen (siehe Grafik). Es kommen zwar jedes Jahr neue Orphan Drugs hinzu, aber es ist auch zu berücksichtigen, dass jedes Jahr für ältere Orphan Drugs die Marktexklusivität abläuft und damit die Förderung endet.
Sondersituation bei der frühen Nutzenbewertung
Im Jahr 2011 wurde mit dem „Arzneimittelmarkt-Neuordnungsgesetz“, kurz AMNOG, ein neues Bewertungsverfahren zur Bewertung innovativer Arzneimittel eingeführt. Dabei überprüft der Gemeinsame Bundesausschuss (G-BA) nach der Zulassung noch einmal gesondert beim Markteintritt, ob ein neuer Wirkstoff einen Zusatznutzen gegenüber der bestehenden Standardtherapie aufweisen kann. Nur dann darf das neue Medikament teurer sein, als die bisherigen Medikamente gegen die jeweilige Krankheit.
Für Orphan Drugs wurde eine Sondersituation geschaffen: Bei ihnen ist bereits mit der Zulassung belegt, dass sie über einen Zusatznutzen verfügen. „Diese Regelung hat gute Gründe“, sagt Wilken.
„Denn entweder kommt mit dem neuen Wirkstoff sowieso eine erstmalige Therapie gegen die jeweilige Seltene Erkrankung auf den Markt, dann gibt es gar keine adäquate Vergleichstherapie. Oder das neue Arzneimittel bringt einen klinisch relevanten Vorteil gegenüber dem bisherigen Orphan Drug. Sonst hätte das bisherige Medikament seine Marktexklusivität, die ihm ja nach der EU-Verordnung zusteht, nicht verloren.“ Der G-BA darf den Zusatznutzen der Höhe nach bemessen und es finden Preisverhandlungen statt. Voraussetzung für diese Regelung ist allerdings, dass das Unternehmen mit diesem Medikament unterhalb der Umsatzschwelle von 50 Millionen Euro pro Jahr (Stand Juni 2022) bei der Gesetzlichen Krankenversicherung bleibt.
Die Orphan-Drug-Sonderregelungen stehen immer wieder in der Diskussion, zumal die Therapien kostspielig sein können. Als „teuerstes Medikament der Welt“ hat zum Beispiel die Gentherapie Zolgensma im Jahr 2020 für Schlagzeilen gesorgt. Doch den Kosten steht ein enormer Nutzen gegenüber. Sowohl für Patientinnen und Patienten als auch für die gesamte Medizin.
„Es ist ein goßartiger Fortschritt, dass mit einer Injektion die Spinale Muskelatrophie behandelbar wird. Kinder, die zuvor nur wenige Jahre lebten und erheblich eingeschränkt waren, überleben und lernen auf einmal laufen und Rad fahren. Wenn der Therapievorteil derart offensichtlich ist, sollte sich die öffentliche Debatte meines Erachtens nicht auf die Kostenseite fokussieren, sondern auf den Nutzen für die Patientinnen und Patienten. Die Frage ist doch, wohin lenken wir Forschungsgelder und die Etats unseres überwiegend solidarisch finanzierten Gesundheitssystems?“, merkt Prof. Annette Grüters-Kieslich von der Eva-Luise- und Horst-Köhler-Stiftung für Menschen mit Seltenen Erkrankungen an.
Natürlich seien Innovationen, wie solche hochpräzisen gentherapeutischen Verfahren, anfangs sehr teuer, doch stehe den Investitionen nicht nur ein individueller Patientennutzen, sondern auch ein allgemeiner Erkenntnisgewinn gegenüber: „Anhand von Seltenen Erkrankungen lernen wir viel für die Behandlung von häufigen Krankheitsbildern und davon profitieren alle.”
Arzneimittelkosten sind stabil
Dr. Kai Joachimsen, Hauptgeschäftsführer des Bundesverbandes der Pharmazeutischen Industrie e.V., betont, dass eine Überlastung der Gesetzlichen Krankenversicherung durch Orphan Drugs nicht gegeben ist. „Die Ausgaben der gesetzlichen Krankenkassen für Arzneimittel sind, bezogen auf das Bruttoinlandsprodukt, seit Jahren stabil bei rund einem Prozent.“ Und er rechnet weiter vor:
„Der Anteil der Arzneimittel am GKV-Gesamtmarkt beläuft sich nach Abzug der Abschläge ebenfalls stabil auf aktuell rund elf Prozent.“ Doch Patientinnen und Patienten hören immer wieder, dass die hohen Preise für die Orphan Drugs zu Lasten der Solidargemeinschaft gehen – ein Argument, das Sabine Biermann, Vorstandsvorsitzende der Gaucher Gesellschaft Deutschland e.V., nur zu Genüge kennt. (s. Fallbeispiel). Sie wünscht sich, dass die Menschen mit Seltenen Erkrankungen nicht aussortiert werden. „Jeder hat ein Anrecht auf eine Therapie“, so Biermann.
Gute Datenlage
Weiterer Kritikpunkt: Der Nutzen sei durch die Erleichterungen bei der Zulassung und im AMNOG nicht ausreichend belegt. Deshalb gehöre der Sonderstatus abgeschafft. „Die Orphan Drugs verfügen in der AMNOG-Bewertung über eine solide Datengrundlage“, hält Dr. Kai Joachimsen, Hauptgeschäftsführer des BPI, entgegen. „Auch bei kleinen Patientenkollektiven gibt es sehr häufig randomisierte klinische Studien.“
Ein solcher Studientyp gilt als Goldstandard der medizinischen Wissenschaft. Die Studienteilnehmenden werden per Zufall unterschiedlichen Untersuchungsgruppen zugeordnet, um verschiedene Medikamente (neues Medikament versus Scheinmedikament bzw. Standardtherapie) miteinander vergleichen zu können. Eine Auswertung der SKC-Beratungsgesellschaft im Auftrag des BPI bestätigt: Selbst bei einer sehr kleinen Teilnehmergröße von weniger als 50 Patienten beträgt der Anteil von randomisierten kontrollierten Studien immerhin noch knapp ein Drittel, bei über 1.000 Patienten liegt der Anteil schon bei 80 Prozent.
Dr. Matthias Wilken vom BPI weist darauf hin, dass man das Studiensetting bei Seltenen Erkrankungen nicht vergleichen könne mit einer Studie zu Diabetes zum Beispiel, weil naturgemäß sehr viel weniger Patienten zur Verfügung stehen. „Man muss die Patientinnen und Patienten manchmal in ganz Europa oder überall auf der Welt zusammensuchen, das ist ein sehr großer Aufwand.“ Oftmals kann man das neue Medikament nicht, wie gefordert, mit einer herkömmlichen Therapie vergleichen, weil die gar nicht existiert. Auch wird immer wieder von Kritikern bemängelt, dass die Studiendauer zu kurz sei.
„Doch längere Studien sind bei lebensbedrohlichen oder rasch fortschreitenden Erkrankungen ethisch oft nicht vertretbar“, so Wilken. Der BPI kritisiert, dass das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen, das im Auftrag des G-BA auch Orphan Drugs bewertet, in Deutschland bisher noch keine Bewertungssystematik entwickelt hat, die den Besonderheiten der Orphan Drugs gerecht wird.
Von den Seltenen für die Häufigen lernen
Auch Prof. Christoph Klein von der Kinderklinik am Universitätsklinikum München, ein Zentrum für Seltene Erkrankungen, kann die Kritik bezüglich einer mangelhaften Datenlage nicht nachvollziehen:
„Gerade bei Orphan Drugs haben wir oft sehr viel mehr Einblicke in die Mechanismen der Wirksamkeit als bei Medikamenten gegen häufige Erkrankungen, bei denen wir oft gar nicht genau wissen, was da im Körper eigentlich passiert.“ Erkrankungen wie Brustkrebs, Alzheimer oder Arteriosklerose gingen mit einer unüberschaubaren Vielfalt an Veränderungen auf zellulärer und molekularer Ebene einher, so Klein. Bei Seltenen Erkrankungen ist dagegen oftmals nur ein einziges Gen betroffen. „Damit können wir das Übel an der Wurzel packen und damit werden wir zunehmend erfolgreicher sein.“
Er verweist auf die Fülle von technologischen Möglichkeiten, die in Zukunft mit der Gentherapie zur Verfügung stehen: Das CRISPR/Cas-System – die sogenannte Genschere, mit der sich Gene ganz gezielt verändern lassen oder das „Gene Silencing“, das krankmachende Gene hemmt beziehungsweise „stumm“ schaltet oder die Therapien mit körpereigenen Zellen des Patienten (siehe Themendienst Arzneimittel-Innovationen). Damit treibt die Forschung an Seltenen Erkrankungen die sogenannte personalisierte Medizin voran und davon profitieren auch die Therapien von häufigen Erkrankungen. Allein deshalb lohne es sich, so Klein, in die Forschung von Ursachen und Therapien von Seltenen Erkrankungen zu investieren. Doch er muss in seinem Berufsalltag immer wieder feststellen: „Insbesondere die Kinder mit Seltenen Erkrankungen sind überhaupt nicht im Fokus der Gesundheitspolitik.“
Die Zahl beeindruckt: 28 Bündnispartner haben sich im Nationalen Aktionsbündnis für Menschen mit Seltenen Erkrankungen, kurz: NAMSE, auf höchster Ebene zusammengeschlossen. Das Aktionsbündnis wurde im Jahr 2010 auf Initiative des Bundesministeriums für Gesundheit (BMG) und der Selbsthilfe-Vereinigung ACHSE e.V. (Allianz Chronischer Seltener Erkrankungen e.V.) gegründet, um die Versorgung der Betroffenen zu verbessern. Mit im Boot sind neben dem BPI zum Beispiel auch die Bundesärztekammer, verschiedene Bundesministerien, die Deutsche Krankenhausgesellschaft, die Kassenärztliche Bundesvereinigung, der Deutsche Pflegerat, der Gemeinsame Bundesausschuss (G-BA) sowie der Spitzenverband Bund der Krankenkassen (GKV). Was hat das NAMSE bisher bewirkt und was ist noch zu tun? Fragen an die Leiterin der Geschäftsstelle, Dr. Miriam Schlangen.
52 Maßnahmen hat das NAMSE im Jahr 2013 in einem Aktionsplan veröffentlicht. Wie sieht es mit der Umsetzung aus?
Zunächst einmal werten wir es als großen Erfolg, dass sich überhaupt alle relevanten Player im Gesundheitswesen auf diese Maßnahmen einigen konnten. Das war ein zähes Ringen! Der Aktionsplan hat unglaublich viele Initiativen angestoßen. Um nur einige Beispiele zu nennen: Ab dem Jahr 2023 haben wir im stationären Bereich eine Kodierung, so dass Seltene Erkrankungen eindeutig identifiziert werden können. Das ermöglicht valide statistische Auswertungen über die Anzahl der Patienten mit einer solchen Erkrankung und die Zuordnung zum zuständigen Fachgebiet. Außerdem: Die Entwick- lung von elf Leitlinien zu Seltenen Erkrankungen wird vom G-BA gefördert, damit die Diagnose und Behandlung auf evidenzbasierten Füßen steht. Es werden krankheitsspezifische und Patientenregister aufgebaut, um zum Beispiel für klinische Studien Patienten rekrutieren zu können. Besonders stolz aber sind wir auf die Einrichtung der Zentren für Seltene Erkrankungen.
Die Gründung von Zentren für Seltene Erkrankungen bildet das Herzstück des Aktionsplans. Geplant sind A-, B- und C-Zentren. Was hat es mit diesen Stufen auf sich?
Die Typ-A-Zentren sind an Universitätskliniken beheimatet und übernehmen eine Lotsenfunktion für Patientinnen und Patienten mit unklarer Diagnose. Dort arbeiten Expertinnen und Experten verschiedener Fachrichtungen krankheitsübergreifend zusammen, betreiben Grundlagen- und klinische Forschung. Alle Typ-A-Zentren – wir haben in Deutschland mittlerweile 37 – sind miteinander vernetzt. Ist eine Diagnose gestellt oder zumindest eingegrenzt, werden die Patientinnen und Patienten an sogenannten B-Zentren weiterbehandelt, das sind krankheitsspezifische Fachzentren. Zentren vom Typ C, die die wohnortnahe ambulante Versorgung sicherstellen sollen, haben wir leider noch nicht.
Wie sieht es mit der Finanzierung der Zentren aus?
Wir sind in großer Sorge, dass einigen Zentren die Luft ausgeht, weil die Frage der Finanzierung noch nicht gelöst ist. Viele Vergütungsinstrumente passen nicht für Seltene Erkrankungen, wie etwa die Abrechnungsziffern der ambulanten spezialfachärztlichen Versorgung. Ob die Hochschulambulanzpauschale bei der Ambulanz eines Zentrums wirklich ankommt, muss sich noch herausstellen. Durch das Projekt Translate NAMSE sind wir einen Schritt weiter: Es konnte gezeigt werden, dass die Maßnahmen aus dem Nationalen Aktionsplan funktionieren. Jetzt muss noch geschaut werden, ob die vorhandenen Vergütungsinstrumente in der Regelversorgung wirklich greifen.
Zum Maßnahmenplan gehören auch innovative Technologien. Was ist im Bereich der genetischen Diagnostik passiert?
Das sogenannte Next Generation Sequencing, kurz NGS, hat Eingang in die Diagnostik Seltener Erkrankungen gefunden. Beim NGS können die Bausteine vieler hunderter Gene parallel analysiert werden, statt wie vorher nacheinander, was die Sequenzierung schneller und kostengünstiger werden lässt. Außerdem ist im Herbst 2021 das Projekt genomDE gestartet, das vom BMG gefördert wird. Das ist ein tolles Projekt, weil hier Forschung und Versorgung zusammengebracht werden. Ziel ist der Aufbau einer bundesweiten Plattform, damit die Genomsequenzierung besser im klinischen Alltag angewendet werden kann. Im kommenden Jahr startet ein Modellvorhaben, das die Genomsequenzierung bei Seltenen Erkrankungen in die Regelversorgung überführen soll, so dass die GKV die Kosten erstattet.
Wie gut ist das Phänomen Seltene Erkrankungen bei niedergelassenen Ärztinnen und Ärzten bekannt? Das sind ja diejenigen, die bei einem Verdacht in ein Zentrum für Seltene Erkrankungen überweisen.
Wir müssen nach wie vor das Bewusstsein dafür schulen, dass es neben den häufigen auch die Selte- nen Erkrankungen gibt. Das ist sicherlich auch eine Frage der Ausbildung. Wir haben es geschafft, dass im medizinischen Ausbildungskatalog die Seltenen Erkrankungen explizit genannt sind. Eine Ärztin, ein Arzt muss und kann nicht alle 8.000 Seltenen kennen, sollte aber bei einer ungewöhnlichen Kombination von Symptomen auf die Idee kommen, dass es sich um eine Seltene Erkrankung handeln könnte. Eine andere Idee ist es, einen Facharzt für Seltene Erkrankungen zu schaffen, das würde die Problematik noch sichtbarer machen.
Woran liegt es noch, dass die wohnortnahe Versorgung nach wie vor ein Problem ist?
Es gibt zum Beispiel häufig Schwierigkeiten bei der Verordnung der Orphan Drugs. Die Ärzte fürchten, dass sie mit den meist hochpreisigen Medikamenten ihr Budget überschreiten. Doch das scheint eher eine psychische, statt faktische Barriere zu sein. Nach einer Beantragung durch die Ärztin/den Arzt übernehmen die gesetzlichen Krankenkassen in der Regel die Kosten, die Ärztinnen und Ärzte müssen keinen Regress fürchten. Tatsache ist allerdings: Wir haben bislang noch viel zu wenig Medikamente. Denn die Orphan Drugs werden, auch wenn sie teuer sind, den Pharmafirmen in der Regel keinen großen Gewinn bringen.

- … rund fünf Prozent der Neugeborenen von einer Seltenen Erkrankung betroffen sind? (Quelle: Eva Luise und Horst Köhler Stiftung für Menschen mit Seltenen Erkrankungen)
- … in anderen Ländern schon länger als in Europa Sonderregelungen für Orphan Drugs existieren? In den USA etwa gibt es den Orphan Drug Act“ schon seit 1983, in Japan die Orphan Drug Legislation“ seit 1993, in Singapur die Orphan Legislation”
- seit 1997 und in Australien seit 1998. (Quelle: BPI)
- … weniger als ein Prozent (0,8 Prozent) der gesamten Ausgaben der Gesetzlichen Krankenversicherung im ambulanten Bereich auf Arzneimittel für Seltene Erkrankungen entfällt? (Quelle: BPI)
- … allein 2020 und 2021 insgesamt 36 neue Orphan Drugs zugelassen wurden? (Quelle: BPI)
- …dass es etwa 40 Seltene Erkrankungen gibt, von dem nur ein Mensch auf der Welt betroffen ist? (Quelle: Orphanet Berichtsreihe)
Letzter Zugriff auf alle Internetseiten: Juni 2022
Aktuelle Informationen zu Orphan Drugs auf den Seiten des BPI:
https://www.bpi.de/de/alle-themen/orphan-drugs#:~:text=Was%20wie%20kindliche%20Phantasie- w%C3%B6rter%20klingt,seltenen%20Erkrankungen%20hei%C3%9Fen%20Orphan%20Drugs
Nationales Aktionsbündnis für Menschen mit Seltenen Erkrankungen:
https://www.namse.de
ACHSE, Allianz Chronischer Seltener Erkrankungen, Selbsthilfe:
https://www.achse-online.de/de/index.php
Orphanet, das Portal für seltene Krankheiten und Orphan Drugs:
https://www.orpha.net/consor/cgi-bin/index.php
Versorgungsatlas für Menschen mit Seltenen Erkrankungen:
https://www.se-atlas.de
Care-for-Rare Foundation für Kinder mit Seltenen Erkrankungen:
https://www.care-for-rare.org
Eva-Luise- und Horst-Köhler-Stiftung für Menschen mit Seltenen Erkrankungen:
https://www.elhks.de
Gaucher Gesellschaft Deutschland e.V.:
https://www.ggd-ev.de
Bundesverband der Pharmazeutischen Industrie (BPI e.V.):
Aktuelle Informationen auf den Seiten des BPI:
https://www.bpi.de/alle-themen/orphan-drugs
BPI-Informationsbroschüre und Kernforderungen zu Orphan Drugs: https://www.bpi.de/de/nachrichten/detail/bpi-informationsbroschuere-zu-orphan-drugs-und-politi- sche-kernforderungen
Analyse der SKC-Beratungsgesellschaft:
https://www.bpi.de/nachrichten/detail/daten-zur-debatte-orphan-drugs
Verband der forschenden Arzneimittelhersteller (vfa e.V.)
Aktuelle Informationen auf den Seiten des vfa:
https://www.vfa.de/de/arzneimittel-forschung/seltene-erkrankungen
Broschüre „Forschung für die Waisen des Gesundheitssystems. Medikamente für Menschen mit seltenen Erkrankungen.“
https://www.vfa.de/download/forschung-fuer-die-waisen-des-gesundheitssystems.pdf
Positionspapier zu Arzneimitteln für neuartige Therapien (ATMP):
https://www.valicare.com/sites/default/files/2021-04/positionspapier-atmp.pdf
Zugelassene Orphan Drugs:
https://www.vfa.de/de/arzneimittel-forschung/datenbanken-zu-arzneimitteln/orphan-drugs-list
Weitere Quellen:
Beschlusstext vom Innovationsausschuss beim G-BA zum Projekt Translate-Namse:
https://innovationsfonds.g-ba.de/downloads/beschluss-dokumente/157/2022-04-01_TRANSLA- TE-NAMSE.pdf
Nationaler Aktionsplan des NAMSE:
https://www.namse.de/fileadmin/user_upload/downloads/Nationaler_Aktionsplan.pdf
Aileen Hohnstein: „Seltenen Erkrankungen auf der Spur“, in: kma 5/21
https://www.kma-online.de/aktuelles/medizin/detail/seltenen-erkrankungen-auf-der-spur-a-45517
„Von den Seltenen für die Häufigen lernen“, Podcast der Ärzte-Zeitung mit Prof. Annette Grüters-Kieslich
https://www.aerztezeitung.de/Kooperationen/Von-den-Seltenen-fuer-die-Haeufigen-lernen-422610.html
Thomas Meißner: „Wie Zentren für Seltene Erkrankungen Ärzten nutzen können“, in: Ärzte-Zeitung: 28. Februar 2022
https://www.aerztezeitung.de/Medizin/Wie-Zentren-fuer-Seltene-Erkrankungen-Aerzten-nutzen-koennen-427011.html
Beilage vom Tagesspiegel zum Tag der Seltenen Erkrankungen am 28. Februar 2022
(nicht im Netz verfügbar)
AWMF-Leitlinie zum Neugeborenen-Screening:
https://www.awmf.org/uploads/tx_szleitlinien/024-012l_S2k_Neugeborenenscreening_2022-02_01.pdf
genomDE: Nationale und Europäische Genominitiativen – Digitale Konferenz am 30. November 2020
https://www.bundesgesundheitsministerium.de/themen/gesundheitswesen/personalisierte-medizin/genomde-de/digitale-konferenz.html
Kritik an Orphan Drugs:
Auswertung des IQWiG von Januar 2022
https://www.iqwig.de/presse/pressemitteilungen/pressemitteilungen-detailseite_58496.html
„Reimann: Ausnahmeregelung für Orphan Drugs abschaffen.“ Ein Statement der AOK vom 25. Februar 2022
https://www.aok-bv.de/positionen/statements/index_25332.html
Ev Tebroke: „Keine Ausnahmen für Orphan Drugs“, in: Pharmazeutische Zeitung vom 14. Januar 2022
https://www.pharmazeutische-zeitung.de/keine-ausnahmen-fuer-orphan-drugs-130744/
„Die 2-Millionen-Spritze“, in: Securvita Krankenkasse 2/20
https://www.securvita.de/fileadmin/inhalt/dokumente/auszuege_SECURVITAL/202002/securvital_0220_26.pdf
Maike Telgheder: „Zwei-Millionen-Gentherapie von Novartis kann bald zugelassen werden“, in: Handelsblatt 27. März 2020
https://www.handelsblatt.com/unternehmen/industrie/zolgensma-zwei-millionen-gentherapie-von-novar- tis-kann-bald-zugelassen-werden/25690516.html