
Arzneimittelinnovationen
Neue Therapien gegen Krebs oder gegen seltene Erkrankungen, eine Impfung gegen das Coronavirus SARS-CoV-2: Unsere Gesundheit hängt unter anderem von der Entwicklung neuer Arzneimittel und Impfstoffe ab.
Doch Innovationen haben ihren Preis, denn die Forschung dauert in der Regel viele Jahre und die Anforderungen an die Qualität sind extrem hoch. Zudem macht es oft eine Vielzahl an Regularien den Pharmaunternehmen schwer, ihre Forschungsanstrengungen zu refinanzieren. Trotzdem ist die Pharmaindustrie die forschungsintensivste Branche Deutschlands – vor Automobil-, Luft- / Raumfahrzeug- und Maschinenbau sowie chemischer Industrie. Welche neuen Ansätze gibt es in der Arzneimittelforschung? Und welche Vorschriften garantieren die Qualität der Forschung und regulieren die Preise? Und wie können Innovationen schneller bei den Patientinnen und Patienten ankommen? Dieser Themendienst gibt einen Überblick.
Wie immer können Sie das Text- und Bildmaterial des Themendienstes gerne unter Nennung der Quellen kostenlos redaktionell verwenden.
Ihr BPI-Presseteam
Erfolgsgeschichte Hepatitis C: Noch vor etwa 25 Jahren lag die Heilungsquote der chronischen Lebererkrankung bei nur fünf bis zehn Prozent. Viele der Patientinnen und Patienten entwickelten eine Leberzirrhose oder Leberkrebs. Dank neuer Medikamente, die seit 2014 verfügbar sind, können mehr als 95 Prozent der etwa 250.000 Betroffenen in Deutschland geheilt werden – und das bei recht wenigen Nebenwirkungen.
„Die neuen Hepatitis-C-Arzneimittel sind ein Beispiel dafür, wie gut Wissenschaftlerinnen und Wissenschaftler das komplexe Innere der menschlichen Zelle inzwischen erforscht haben“, berichtet der Biologe Dr. Pablo Serrano, Geschäftsfeldleiter Innovation & Forschung sowie Biotechnologie beim Bundesverband der Pharmazeutischen Industrie (BPI) e.V.. Die neuen Medikamente – Direct Acting Antiviral Agents, kurz DAA genannt – wirken direkt in den Leberzellen und blockieren dort gezielt einzelne Schritte der Virusvermehrung, indem sie bestimmte Eiweiße und Enzyme hemmen.
Durch Erkenntnisse der Zell- und Molekularbiologie, Biochemie und Immunologie verstehen Forscherinnen und Forscher die Vorgänge in den Körperzellen immer besser: Wie und wo stellen die Zellen welche Proteine, also Eiweißstoffe, her? Wie teilen sie sich? Wie setzen sie Reparaturen in Gang? Wie kommunizieren sie miteinander? Wie entsorgen sie den Zellmüll? Und wie missbrauchen Viren die Produktionsmaschinen in menschlichen Zellen, um immer neue Viren zu bilden? Dabei dringt die Forschung bis in den Kern der Zelle vor, wo sich die Gene befinden. Mit der Entschlüsselung des Genoms können Wissenschaftlerinnen und Wissenschaftler der Frage nachgehen: Welche Gene können krank machen und wie können krank machende Vorgänge gestoppt, stillgelegt oder repariert werden (s. Gentherapien)? Durch all diese Erkenntnisse wirken neue Arzneimittel immer gezielter. Dabei hat sich der Blickwinkel in der Arzneimittelforschung verändert, erklärt Dr. Serrano. „Bildlich gesprochen stehen Forscher nicht mehr vor einer verschlossenen Tür und müssen durch ein Schlüsselloch in das Innere der Zelle schauen, um die krank machenden Vorgänge zu erspähen. Nun stehen sie oft auf der anderen Seite, hinter der Tür, begehen und erkunden die Zelle und versuchen von dort aus, passende Werkzeuge zu entwickeln, um in das Zellgeschehen gezielt einzugreifen.“
Im Jahr 2019 haben 25 solcher neuen maßgeschneiderten Medikamente mit neuen Wirkstoffen eine Zulassung bekommen. Davon richten sich zehn gegen Krebserkrankungen (siehe unten) und sieben gegen seltene Erkrankungen (siehe unten). Der Verband Forschender Arzneimittelhersteller e.V. (vfA) rechnet damit, dass bis spätestens Ende 2023 weitere innovative Arzneimittel gegen 145 meist schwere Erkrankungen neu zugelassen werden.
Es muss nicht immer ein neuer Wirkstoff mit neuem Wirkmechanismus sein:
Auch Medikamente auf Basis eines bereits bewährten Wirkstoffs können Innovationen sein, sogenannte Schrittinnovationen – im Unterschied zu Sprunginnovationen mit einem neuen Wirkstoff. „Kleine Schritte können oftmals ebenso wertvoll sein wie große Sprünge und tatsächliche Verbesserungen für Patientinnen und Patienten bringen“, sagt Dr. Serrano. Dabei kann es sich zum Beispiel um eine neue Darreichungsform eines altbekannten Medikaments handeln: Eine zwei Millimeter große Minipille eines Herzmedikaments etwa, dass damit auch herzkranken Säuglingen gegeben werden kann, oder ein Medikament in Form eines Pflasters, das den Wirkstoff über die Haut abgibt und erst nach einigen Tagen gewechselt werden muss – eine Lösung für Demenzerkrankte, die oft Schluckprobleme haben. Bei der Parkinson-Erkrankung zum Beispiel können transdermale Pflaster dazu beitragen, motorische Komplikationen zu vermeiden. Auch solche Weiterentwicklungen brauchen pharmazeutische Forschung und klinische Studien: die neue Darreichungsform, die neue Indikation muss getestet werden, eventuell auch an einer besonders verletzlichen, vulnerablen Patientengruppe, etwa Kindern.
Der Schwerpunkt der Arzneimittelinnovationen liegt im Bereich der Onkologie. „Krebszellen sind Saukerle“, betont der Pharmazeut Prof. Theo Dingermann, Seniorprofessor für Pharmazeutische Biologie an der Universität Frankfurt (s.
Interview). „Sie vermehren sich unkontrolliert und können sich vor dem Immunsystem verstecken.“ Kein Wunder, dass Forscherinnen und Forscher sich darum bemühen Wege zu finden, wie man diese heimtückischen Zellen gezielt ausschalten kann. Zehn der 25 neu zugelassenen Medikamente im Jahr 2019 richten sich gegen Krebserkrankungen. Und nach Zahlen des Verbands Forschender Arzneimittelhersteller e.V. (vfA) beschäftigen sich von 434 Projekten der forschenden Pharma-Unternehmen, die voraussichtlich bis Ende 2023 zu einer Zulassung eines neuen Medikaments führen, knapp die Hälfte mit Krebstherapien. Operation, Bestrahlung, Chemotherapie – dieser Dreiklang der Krebstherapie hat aber deshalb nicht ausgedient. „Neue Ansätze, wie zielgerichtete Therapien, die CAR-T-Zell-Therapie oder Immuntherapien sind als Ergänzungen der klassischen Krebstherapien zu verstehen“, betont Prof. Dingermann.
Mehr als Schrotschüsse: zielgerichtete Therapien
Eine Chemotherapie greift alle Zellen an, die sich besonders schnell vermehren. Denn die Medikamente, so genannte Zytostatika, stören ganz allgemein die Zellteilung. So werden Krebszellen zerstört, aber auch andere gesunde Zellen, die sich häufig teilen, wie etwa die Zellen der Haarwurzel. Das erklärt, warum vielen Patientinnen und Patienten während der Behandlung die Haare ausfallen. „Auch wenn der Nutzen einer Chemotherapie klar belegt ist, kann man sie getrost als ‚Schrotschusstherapie‘ bezeichnen“, so Dingermann.
Um Nebenwirkungen in Grenzen zu halten und Krebszellen gezielter zu zerstören, arbeiten Forscherinnen und Forscher an Medikamenten, die spezifischer wirken als eine Chemotherapie. Sie sollen sich nur auf Krebszellen mit bestimmten molekularbiologischen Eigenschaften stürzen und damit gesundes Gewebe schonen. Diese neuen molekularbiologischen Therapien nennen sich auch „zielgerichtete Therapien“ (engl. “Targeted Therapy“).
Diese Arzneimittel fangen zum Beispiel bestimmte Botenstoffe ab, über welche die Zelle bestimmte Befehle bekommt, etwa sich zu vermehren. Oder die Medikamente besetzen bestimmte „Antennen“ an der Zelloberfläche (Rezeptoren genannt), an die die Botenstoffe andocken. Oder sie behindern bestimmte Signalwege im Inneren der Zelle, wo die Signale an den Zellkern weitergegeben werden.
Solche Therapien können die Teilung und das Wachstum von Krebszellen unterdrücken. Oder sie stören die Bildung neuer Blutgefäße, die die Tumorzellen für ihr Wachstum brauchen. Oder sie hemmen bestimmte Enzyme, so dass zum Beispiel das Selbstmordprogramm der Krebszellen gefördert oder die Müllentsorgung blockiert wird, so dass sie an ihrem eigenen Abfall ersticken.
Bei den Medikamenten handelt es sich um molekularbiologisch hergestellte Antikörper, sogenannte monoklonale Antikörper (Wirkstoffname endet auf „-mab“: monoclonal antibodies), die passgenau an Rezeptoren an der Zelloberfläche binden können. Oder es sind Wirkstoffe, die in die Tumorzelle eindringen, sogenannte Kinasehemmer (Wirkstoffname endet auf „-mib“ oder „-nib“). Kinasen sind Eiweiße, die Signale übertragen, die dazu führen, dass die Krebszelle wächst und sich teilt.
Zukunft: Krebstherapie unabhängig vom betroffenen Organ
Doch nicht für jede Krebsart stehen zielgerichtete Therapien zur Verfügung. „Ob eine solche Therapie in Frage kommt, hängt aber nicht nur von der Krebsart ab, sondern auch davon, ob die jeweiligen Zielstrukturen auf oder in den Krebszellen vorhanden sind“, betont Dingermann. Deshalb ist bei einigen Krebspatienten ein diagnostischer Test dem Beginn einer Behandlung vorgeschaltet, um herauszufinden, ob sie diese „Biomarker“, also diese genetischen, molekularen oder zellulären Besonderheiten der Tumorzellen aufweisen. Denn nur dann können die Patienten von den zielgerichteten Medikamenten profitieren. „In Zukunft wird eine Krebstherapie nicht mehr organspezifisch erfolgen, sondern abhängig von Biomarkern – egal, ob es sich um Brust-, Darm-, Prostata- oder Lungenkrebs handelt“, so Dingermann. Ein erstes Medikament für jede Krebserkrankung, die unabhängig vom betroffenen Organ mit einer bestimmten Genmutation in Verbindung steht, wurde 2019 zugelassen.
„Scharfmacher“: Immuntherapien
Die Immuntherapien in der Krebstherapie bedeuten einen „Paradigmenwechsel“, so Dingermann: „Denn nicht mehr der Tumor wird behandelt, sondern das Immunsystem soll den Job übernehmen.“ Eigentlich sollte das Immunsystem bösartig veränderte Zellen im Körper erkennen, doch Tumorzellen können dem Immunsystem auf verschiedenen Wegen ausweichen. Deshalb forschen Wissenschaftlerinnen und Wissenschaftler intensiv daran, wie sich diese Ausweichmechanismen der Krebszellen gezielt ausschalten lassen.
Bei der CAR-T-Zell-Therapie werden bestimmte Immunzellen des Patienten, nämlich die T-Zellen, mit gentechnischen Mitteln „scharf gemacht“. Dafür werden die T-Zellen dem Patienten entnommen und im Labor mit einem zusätzlichen Gen ausgestattet, so dass sie einen Zapfen, also Rezeptor ausbilden, der die Krebszellen als Feind erkennen kann. Diese gentechnisch veränderten T-Zellen können gezielt an die Krebszellen andocken und sie abtöten. „Bestimmte Formen der Leukämie lassen sich damit sehr effektiv bekämpfen“, berichtet Prof. Dingermann. 2018 sind erstmals zwei CAR-T-Zell-Therapien gegen spezielle Krebserkrankungen des Bluts und des Lymphsystems zugelassen worden. Die CAR-T-Zell-Therapie wird als Gentherapie eingestuft und ist sozusagen ein „lebendes Medikament“, denn sie besteht aus lebenden Immunzellen des Patienten.
Pfiffig ist auch der Ansatz der Immun-Checkpoint-Hemmer: Im Immunsystem sind sogenannte Immun-Checkpoints eingebaut, also Kontrollpunkte, die die eigene Immunabwehr bremsen. Tumorzellen aktivieren diese Checkpoints, indem sie bestimmte Eiweiße bilden – das Immunsystem wird also heruntergefahren und kann die Krebszellen nicht bekämpfen. Die Immun-Checkpoint-Hemmer schalten diese Eiweiße aus, so dass die Blockierung der Immunabwehr aufgehoben wird und der Tumor angegriffen werden kann. Immun-Checkpoint-Hemmer sind bereits bei einigen Krebsarten wie Lungenkrebs, schwarzem Hautkrebs oder Blasenkrebs zugelassen.
Ein Drittel aller Medikamente mit einem neuen Wirkstoff, die in Deutschland zwischen 2014 und 2018 herauskamen, sind sogenannte “Orphan Drugs“. Das englische Wort “orphan“ heißt „Waise“ und bedeutet in diesem Zusammenhang, dass es sich um Medikamente handelt, die sich gegen seltene Erkrankungen richten, “Orphan Diseases“ genannt. Orphan Diseases sind dadurch definiert, dass nicht mehr als fünf von 10.000 EU-Bürgern an der jeweiligen Erkrankung leiden. Es sind etwa 8.000 verschiedene seltene Leiden bekannt, darunter bestimmte Krebsformen, Autoimmunkrankheiten, Nerven-, Stoffwechsel- oder Augenerkrankungen. Viele der rund vier Millionen Betroffenen in Deutschland sind Kinder. Im Jahr 2019 haben sieben Medikamente gegen seltene Erkrankungen die Zulassung geschafft. Doch es gibt noch viel zu tun: Rund 95 Prozent der seltenen Erkrankungen müssen noch weiter erforscht werden, um ein Arzneimittel gegen sie zu finden.
Gentherapien
Bei vier von fünf dieser Erkrankungen handelt es um einen Defekt eines Gens. So ist es kein Wunder, dass seltene Erkrankungen das klassische Feld der Gentherapie sind. Nach 50 Jahren Grundlagenforschung wurde 2012 das erste Gentherapeutikum in Europa zugelassen, gegen eine seltene Fettstoffwechselerkrankung. Bei den bisher zugelassenen Gentherapien wird ein neues Gen in den Organismus eingeschleust, und zwar mit Hilfe von „Genfähren“. Das können zum Beispiel unschädlich gemachte Viren sein, die – mit einem Gen bestückt – in die Körperzellen bis zum Zellkern vordringen.
Überblick zugelassene Gentherapien
Bei allen bisher zugelassenen acht Gentherapien handelt es sich um seltene Erkrankungen, darunter auch seltene Krebsarten:
- 2012: erstes zugelassenes Gentherapeutikum in Europa, gegen eine seltene, angeborene Fettstoffwechselkrankheit (Lipoproteinlipase-Mangel), das Medikament ist allerdings nicht mehr auf dem Markt
- 2015: Gentherapeutikum gegen bestimmte Melanome (schwarzer Hautkrebs)
- 2016: Gentherapie gegen einen schweren Immundefekt bei Kindern
- 2018: zwei CAR-T-Zell-Therapien zur Behandlung bestimmter Blutkrebsarten
- 2018: Gentherapie zur Behandlung der Leberschen kongenitalen Amaurose, eine erbliche Augenerkrankung
- 2019: Gentherapeutikum gegen die seltene Bluterkrankung Beta-Thalassämie
- 2020: Gentherapeutikum gegen Spinale Muskelatrophie, eine seltene neurologische Erkrankung bei Kleinkindern.
Das Schweizer Messer: CRISPR/Cas
Mit der Entdeckung der „Gen-Schere“ könnte die Gentherapie eine neue Richtung nehmen. Bei einer der jüngst entdeckten „Werkzeuge“ dieser Art handelt es sich um das sogenannte CRISPR/Cas-System, das eigentlich Bakterien zur Abwehr von Viren nutzt. „Das ist eine sehr scharfe Klinge, mit der Gene ganz gezielt verändert werden können“, sagt Dr. Pablo Serrano vom BPI. Diese Genchirurgie wird zurzeit in klinischen Studien getestet, gegen eine seltene Bluterkrankung, gegen eine seltene, zur Erblindung führenden Augenerkrankung und zur Bekämpfung von HIV.
Gen-Silencing
Unwillkürliche, unkoordinierte Bewegungen – das ist ein Kennzeichen der seltenen erblichen Erkrankung Chorea Huntington, früher deshalb auch Veitstanz genannt. Die Erkrankung beruht auf einem Defekt des Huntington-Gens, was unaufhaltsam zum Untergang von Gehirnzellen führt. Bis heute ist die Erkrankung nicht heilbar, aber ein neuer Ansatz macht Hoffnung: Das sogenannte Gen-
Silencing. „Dabei wird das krankmachende Gen praktisch stillgelegt, indem die Informationen für die Bauanleitung eines fehlerhaften Proteins vor deren Herstellung abgefangen und neutralisiert werden“, so Dr. Serrano.
Basis dieser Technologie ist RNA – das Schwestermolekül der DNA und dafür zuständig, dass die genetische Information aus dem Zellkern heraustransportiert und an die Orte der Proteinproduktion in der Zelle gebracht wird. RNA macht sozusagen eine Abschrift der Gene. Beim Gen-Silencing werden synthetisch hergestellte RNA-Fragmente gespritzt, die sich in den Zellen mit einer bestimmten
natürlichen RNA verbinden, so dass diese natürliche RNA die Information der Gene nicht mehr weitergeben kann und somit die Produktion des fehlerhaften Proteins unterbleibt. Diese Technologie wird deshalb auch RNAi genannt – dabei steht das i für Interferenz also Unterbrechung. Bisher gibt es erst vier zugelassene Medikamente dieser Art. Es wird sich zeigen, ob sich mit diesem neuen Ansatz
auch das Huntington-Gen stumm schalten lässt.
Zwei Millionen Dollar: Das kostet das bisher teuerste Medikament der Welt, das Gentherapeutikum Zolgensma. Es soll die spinale Muskelatrophie heilen, eine seltene Erkrankung. Und gleichzeitig die häufigste genetisch bedingte Todesursache bei Kleinkindern. Weil das Gen SMN1 defekt ist, arbeiten die Nervenzellen im Rückenmark nicht richtig, so dass die Kinder Muskelschwund entwickeln und daran sterben. Die bisherigen Daten zeigen: Nach der Gabe von Zolgensma können die Kinder wieder den Kopf heben, selbstständig sitzen, essen und atmen – Langzeitstudien fehlen allerdings noch.
Auch wenn ein Arzneimittel Leben rettet – darf es deshalb sehr teuer sein? Um solche, oft fälschlicherweise als Mondpreise bezeichneten Preise zu verstehen, hilft ein Blick auf die Arzneimittelforschung: Mehr als zehn Jahre dauert es von einer Idee bis zur Zulassung, manchmal auch zwanzig Jahre. „Dabei können Kosten in Höhe von mehr als einer Milliarde Euro je Substanz entstehen und das bei geringen Erfolgsquoten“, betont der Apotheker Dr. Matthias Wilken, Geschäftsführer Market Access, Märkte und Versorgung beim BPI. Von rund 10.000 Molekülen, die am Anfang als Wirkstoff in Frage kommen, schafft es in der Regel gerade einmal eine Substanz durch den langwierigen Prozess der Erforschung und Zulassung. „Viele Entwicklungen schlagen fehl – diese Kosten müssen auch berücksichtigt werden, das vergessen viele“, so
Dr. Wilken.
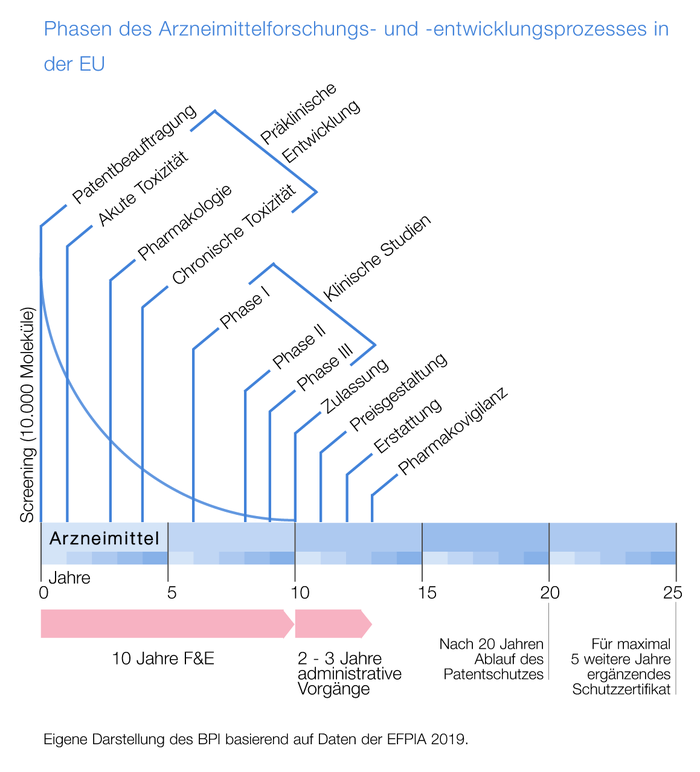
Abbruchkante: Welcher Kandidat schafft es in die klinische Forschung?
Berühmt-berüchtigt ist das sogenannte “Valley of Death“. Das „Tal des Todes“ beginnt mit der „Abbruchkante“ zwischen Grundlagenforschung und klinischen Studien und erstreckt sich über die Studienphasen I bis III (s. Kasten). Bevor eine Substanz in klinischen Studien am Menschen getestet werden kann, stehen viele Jahre Arbeit im Labor an: Die Wirkstoffkandidaten müssen in Zellkulturen und Tiermodellen zunächst beweisen, dass sie eine bestimmte Wirksamkeit haben und vor allem nicht giftig oder unverträglich sind. Haben die Kandidaten diese ersten präklinischen Runden überstanden, geht es eigentlich erst richtig los. Es folgen verschiedene Studienphasen mit gesunden und erkrankten Menschen, um zu testen, wie sich die Substanz im menschlichen Körper verhält, wie verträglich und sicher sie ist und welche Dosierungen eine Wirksamkeit mit möglichst wenig Nebenwirkungen garantieren. Doch diesen Sprung von der präklinischen in die klinische Phase schaffen viele Substanzen nicht. „Neben dem natürlichen Scheitern von Forschungsansätzen ist es manchmal auch eine Frage der Förderung. Die staatlichen Förderprogramme beschränken sich häufig auf die frühen Phasen. Die großen klinischen Studien – die teuersten Phasen in der Forschung – werden in der Regel nicht gefördert“, bemerkt Dr. Wilken.
Nach einer Zulassung gehen Aufwand und Kosten weiter: So steht etwa die Frühe Nutzenbewertung bevor (siehe unten), zudem sind die pharmazeutischen Unternehmen dazu verpflichtet, in der Anwendung Neben- und Wechselwirkungen des Medikaments zu sammeln und auszuwerten, Pharmakovigilanz genannt. Und gerade bei innovativen Arzneimitteln ist eine Zulassung häufig daran geknüpft, weitere Studien nachzureichen.
Vier klinische Phasen
Jedes Medikament muss auf Wirksamkeit, Verträglichkeit und pharmazeutische Qualität hin geprüft werden. Klinische Studien mit freiwilligen Teilnehmerinnen und Teilnehmern erfolgen regelhaft in vier Phasen:
Phase I: Erste Anwendung am Menschen, in der Regel mit wenigen gesunden Erwachsenen, um Verträglichkeit und Sicherheit der Substanz zu testen und zu untersuchen, wie sich die Substanz im Körper verhält und wie sie verstoffwechselt wird (Pharmakodynamik und Pharmakokinetik).
Phase II: In der Regel mit 100 bis 500 Patientinnen und Patienten, um zu prüfen, ob und wie die Substanz bei Erkrankten wirkt und wie verträglich sie ist. Zudem geht es um die richtige Dosierung.
Phase III: In der Regel mit mehreren hundert bis wenigen tausend Patientinnen und Patienten. In dieser Phase muss die Wirksamkeit und Sicherheit an vielen unterschiedlichen Menschen zuverlässig nachgewiesen werden – und zwar im Vergleich mit einem Scheinmedikament (Placebo) oder mit einer Vergleichstherapie. Solche kontrollierten Studien sind meist Grundlage für eine Zulassung bei klassischen Medikamenten und Indikationen.
Phase IV: Studien nach der Zulassung, die die Sicherheit noch über die Zulassung hinaus prüfen sollen (PASS, Post-authorisation Safety Study) oder die Wirksamkeit (PAES, Post-authorisation Efficacy Study). Diese Studien werden manchmal von den Zulassungsbehörden gefordert oder vom pharmazeutischen Hersteller bzw. Inhaber der Zulassung des Arzneimittels freiwillig durchgeführt.
Hoher Forschungsaufwand und häufig nur wenige Patienten: Für die pharmazeutischen Unternehmen ist es – entgegen der öffentlichen Meinung – nicht einfach, ihre Kosten für die Entwicklung eines neuen Arzneimittels zu refinanzieren. Der Gesetzgeber hat viele Instrumente zur Kostendämpfung eingebaut, wie etwa Zwangsabschläge, Festbeträge, Rabattverträge mit den Krankenkassen, Preisstopps oder die Frühe Nutzenbewertung. So ist praktisch jedes Arzneimittel preisreguliert. Nur im ersten Jahr nach der Zulassung erstatten die gesetzlichen Krankenkassen den Preis für ein innovatives Arzneimittel auf der Basis der Kalkulation des Herstellers, doch auch dieser Preis muss am Markt bestehen. „Es ist verständlich, dass die Unternehmen im ersten Jahr den Preis für die Arzneimittelinnovation möglichst hoch ansetzen, um auf ihre Kosten zu kommen“, so Prof. Theo Dingermann. „Anschließend wird der Preis sowieso oft massiv gedrückt.“
Frühe Nutzenbewertung: Zusatznutzen ja oder nein?
Seit das „Arzneimittelmarkt-Neuordnungsgesetz“, kurz AMNOG, im Jahr 2011 in Kraft getreten ist, wird in Deutschland jedes Arzneimittel mit einem neuen Wirkstoff nach der Zulassung noch einmal gesondert durch den Gemeinsamen Bundesausschuss (G-BA), ein Gremium der Selbstverwaltung, geprüft. Hat der Neuling einen Zusatznutzen gegenüber der bestehenden Standardtherapie? Nur dann darf das neue Medikament teurer sein, als die bisherigen Medikamente gegen die jeweilige Krankheit. Aber auch dann können die pharmazeutischen Unternehmen den Preis nicht frei festlegen, sondern müssen mit den Spitzenverband der Gesetzlichen Krankenkassen (GKV-SV) verhandeln.
Das Problem dabei: „Die Standardtherapie ist häufig sehr preiswert und kostet unter Umständen nur einen Euro pro Tag oder noch weniger. Falls die Studien keinen Mehrwert belegen können – was oftmals auch durch die methodischen Vorgaben des Bewertungsverfahrens oder eine abweichende Vorgabe der Vergleichstherapie durch den G-BA bedingt ist – können die pharmazeutischen Unternehmen die Entwicklungskosten nicht im Geringsten einspielen“, gibt Dr. Matthias Wilken vom BPI zu bedenken. Und selbst wenn sich ein Zusatznutzen zeigt, kann das neue Präparat manchmal nicht wirtschaftlich angeboten werden. Denn dann wird mit den Krankenkassen nur über einen Aufschlag auf die möglicherweise sehr billige Vergleichstherapie verhandelt. „Bei bestimmten insbesondere chronischen Erkrankungen, wie Diabetes oder Epilepsie, gibt es deshalb kaum noch Innovationen, obwohl diese benötigt werden“, berichtet Dr. Wilken. „Es lohnt sich dann einfach nicht für die pharmazeutischen Unternehmen.“
Schrittinnovationen (siehe dort) – also Innovationen auf Basis bewährter Wirkstoffe – kommen in der Regel gar nicht erst in die Frühe Nutzenbewertung, sondern werden automatisch in eine Festbetragsgruppe eingegliedert. Das heißt, die gesetzlichen Krankenkassen übernehmen nur Preise bis zu dem jeweiligen Festbetrag. Verordnet der Arzt ein teureres Medikament, muss der Patient die Differenz selber bezahlen. „An dieser Stelle müssen wir über ein erweitertes Verständnis von ‚Innovation‘ mit der Politik diskutieren“, so Dr. Wilken. „Denn nicht nur komplett neue, auch therapeutisch verbesserte Medikamente müssen auf dem Markt die Möglichkeit haben, angemessen höhere Preise zu erzielen, als die Standardpräparate.“
Besondere Regeln für Orphan Drugs
Zumindest für Orphan Drugs haben sich die wirtschaftlichen Rahmenbedingungen verbessert. Denn bei seltenen Erkrankungen sind die Patientenzahlen so klein, dass die Forschungs- und Entwicklungskosten allein deshalb nicht eingespielt werden können. Im Jahr 2000 hat die Orphan Drug-Verordnung in der EU Anreize für die Forschung in diesem Bereich geschaffen: Das neue Medikament ist zehn Jahre lang nicht nur vor Nachahmerpräparaten geschützt, sondern auch davor, dass andere Medikamente gegen die gleiche Krankheit zugelassen werden, die nicht besser sind. Es gibt also ein Jahrzehnt lang eine Marktexklusivität. Das AMNOG hat im Jahr 2011 dem besonderen Status dieser Arzneimittel Rechnung getragen: Auch Orphan Drugs müssen die Frühe Nutzenbewertung durchlaufen, doch die Unternehmen können sich im Regelfall auf das Zulassungsverfahren als ausreichenden Nachweis für einen Zusatznutzen berufen. „Dies ist auch nachvollziehbar, denn entweder ist mit dem Arzneimittel erstmalig überhaupt eine Therapieoption für die betroffenen Patienten vorhanden“, erklärt Dr. Wilken. „Oder die Zulassungsbehörde überprüft beim Vorhandensein von alternativen Behandlungsoptionen, dass das neue Arzneimittel einen ‚signifikanten klinischen Vorteil‘ bietet. Diese Bewertung braucht dann der G-BA nicht zu wiederholen.“ Seit dem Jahr 2000 bis Ende 2018 haben rund 160 Orphan Drugs gegen rund 130 seltene Krankheiten die Zulassung in der EU erhalten. Dabei ist zu beachten, dass ein Orphan Drug seinen Status und die Marktexklusivität in jedem Fall nach spätestens zehn Jahren verliert, auch wenn es danach weiterhin im Markt verfügbar ist.
Offiziell gibt es das neue Coronavirus, genauer SARS-CoV-2, seit dem 31. Dezember 2019. Binnen vier Monaten, also bereits im April 2020, starteten nach der vorklinischen Forschungsphase die ersten Studien für die Suche nach einem Impfstoff am Menschen (s. Kasten). „Damit handelt es sich um die schnellste Impfstoffentwicklung aller Zeiten“, betont Dr. Jens Peters, Geschäftsfeldleiter Klinische Forschung beim BPI. „Üblicherweise dauert das sechs bis zehn Jahre.“ Das sollte jedoch nicht bedeuten, dass Sicherheitsstandards heruntergeschraubt werden, so Dr. Peters. Schließlich müssten Wirkung und Nebenwirkungen bei Impfstoffen besonders gut geprüft sein, da sie für unter Umständen Millionen gesunde Menschen vorgesehen sind. „Mit Blick auf den hohen medizinischen Bedarf bearbeiten die Zulassungsbehörden wissenschaftliche Unterlagen zum Coronavirus vorrangig, Fristen verkürzen sich, auf Gebühren für Beratung und Zulassung wird verzichtet, normalerweise nacheinander folgende Studienphasen können teilweise parallel anlaufen (siehe Interview).
Überall auf der Welt läuft die Corona-Forschung auf Hochtouren. Etwa 150 Impfstoff-Projekte sind in der Pipeline – in der Hoffnung, dass einzelne Kandidaten auch die schwierige dritte Studienphase überstehen, in der ein Impfstoff an vielen Hunderten oder Tausenden Menschen auf Wirksamkeit und Sicherheit geprüft wird. „Dabei bündelt man alle Kräfte: Kommerzielle und akademische Forschung, Behörden und Start-up-Unternehmen arbeiten weltweit verstärkt zusammen“, berichtet Dr. Peters. Auch pharmazeutische Unternehmen kooperieren miteinander, indem sie zum Beispiel Laborkapazitäten oder Sammlungen von Molekülen zur Verfügung stellen.
Das Spike-Protein: Wie Zacken einer Krone
Das Coronavirus, genauer SARS-CoV-2, ist wie alle Viren ein „Parasit“. Es braucht einen Wirt, um sich zu vervielfältigen. So bemächtigt sich das Virus der menschlichen Zellen und zwingt ihnen sein Vermehrungsprogramm auf. Für die Entwicklung eines Impfstoffs und auch eines Arzneimittels gegen die Krankheit COVID-19 ist es also ausschlaggebend zu wissen, wie das Virus in die Zelle eindringt und sich dort vermehrt. Im Fokus der Forscher steht häufig das sogenannte Spike-Protein – ein Eiweiß, mit dessen Hilfe das Virus in die menschliche Zelle eindringen kann. Dieses Protein verleiht dem Virus auch seinen Namen: Wie Zacken einer Krone (Krone = lat. corona) steht es von der Virushülle ab. Weil es so auffällig ist, eignet es sich auch bevorzugt als Angriffspunkt für das Immunsystem. Folgende neuen Ansätze befassen sich damit, wie das Immunsystem gegen das Coronavirus mobilisiert werden kann und wie sich das Spike-Protein dafür nutzen lässt.
Verkleidetes Virus: Vektor-Impfstoffe
Bei diesem neuen Ansatz wird ein harmloses Virus mit gentechnischer Hilfe als SARS-CoV-2 „verkleidet“. Das Virus, meist ein Erkältungsvirus (gehört zur Gruppe der Adenoviren), bildet auf seiner Oberfläche das Spike-Protein des SARS-CoV-2-Erregers aus. Die Geimpften sollen darauf reagieren, indem ihr Immunsystem dieses Protein erkennen und in der Folge das Virus bekämpfen kann. In ersten Studienergebnissen konnten chinesische Forscherinnen und Forscher zwar nachweisen, dass die Teilnehmer Antikörper gebildet haben. Ob die Menschen dadurch wirklich geschützt sind, ist noch unklar.
Der Körper produziert Impfstoff selbst: genbasierte Impfstoffe
Der neueste Typ von Impfstoffen sind genbasierte Impfstoffe, bisher gibt es noch keine zugelassenen Impfstoffe dieser Art. Dabei enthalten die Impfstoffe ausgewählte Gene des Virus. Das geniale Prinzip dahinter: Diese Impfstoffe liefern den Bauplan des Virus, so dass nach der Injektion der menschliche Organismus (ungefährliche) Eiweißstoffe des Virus – darunter auch Spike-Proteine – herstellt. Das Immunsystem reagiert darauf und baut einen Immunschutz wie bei konventionellen Impfstoffen auf. Bei den genbasierten Impfstoffen produziert der Körper also den Impfstoff selbst, während bei den herkömmlichen Tod- oder Lebendimpfstoffen abgetötete oder abgeschwächte Viren bzw. Virenbestandteile gespritzt werden. Basis der neuen Impfstoffe ist vor allem RNA, eine Art Kopie der DNA, also des genetischen Materials im Zellkern. Dabei handelt es sich genauer um eine Boten-RNA (mRNA), die die genetische Information aus dem Zellkern heraustransportiert. Unschlagbarer Vorteil der mRNA-Impfstoffe: Sie können sehr schnell in großen Mengen produziert werden. Die Bundesregierung schätzt diesen Ansatz als so vielversprechend ein, dass sie 300 Millionen Euro in ein süddeutsches Biotech-Unternehmen investiert. Dabei sieht sie auch das Potenzial für mRNA-Impfstoffe gegen andere Infektionskrankheiten oder auch für mRNA-basierte Arzneimittel (s. Antikörper, unten).
Medikamente gegen COVID-19
Auch bei der Suche nach Medikamenten, welche die von dem Coronavirus ausgelöste Krankheit COVID-19 heilen oder lindern können, drücken Forscherinnen und Forscher weltweit aufs Tempo. Zurzeit laufen etwa 1.500 Therapiestudien (also Studien mit Wirkstoffen im Gegensatz zu Studien mit Impfstoffen). Folgende Ansätze lassen sich unter anderem unterscheiden:
- Antivirale Medikamente: Dabei handelt es sich um zum Teil schon zugelassene Medikamente gegen andere Erkrankungen, das sind also Schrittinnovationen. Ein bekannter Kandidat ist das ursprünglich gegen Ebola entwickelte Remdesivir, das die Virusvermehrung im Zellinneren hemmt. Ein weiterer guter alter Bekannter ist zum Beispiel Ivermectin, ein Antiparasitikum, das gegen Fadenwürmer und Krätzmilben wirkt. Auch das eigentlich gegen akutes Lungenversagen entwickelte APN01 gehört in diese Gruppe. Es bindet als „falscher Rezeptor“ an das Spike-Protein am Virus, so dass es nicht mehr an den „richtigen Rezeptor“ an der menschlichen Zelle andocken kann.
- Immunmodulatoren: Bei schweren Verläufen von COVID-19 ist es irgendwann nicht mehr das Virus, sondern das eigene Immunsystem, das den Patienten krank macht. Dämpfende Immunmodulatoren regulieren eine exzessive Immunreaktion herunter und werden auch gegen Autoimmunerkrankungen wie Rheuma oder MS eingesetzt. Bei schwerstkranken COVID-19-Patienten konnte in einer Studie zum Beispiel der Wirkstoff Dexamethason, mit dem allergische Reaktionen, Asthma oder rheumatoide Arthritis behandelt werden, die Sterblichkeit deutlich senken. Auch hier handelt es sich um Innovationen auf Basis bewährter Wirkstoffe, also Schrittinnovationen (s. dort).
- Antikörper: Eigentlich eine alte Methode: Patienten bekommen als Therapie Antikörper gegen SARS-CoV-2-Viren aus dem Blutserum von Menschen, die die Infektion bereits überstanden haben. Die Antikörper sind in der Lage, die Viren im Körper zu neutralisieren. Gentechnisch weitergedreht ist der Ansatz von Antikörpern in Form von mRNA (siehe mRNA-Impfstoff). Injiziert man diese genbasierten Antikörper, stellen die Patienten diese Antikörper für eine Weile selber her.
- Gen-Silencing: Zwei US-Unternehmen und ein südkoreanisches arbeiten an diesem ganz neuen Ansatz: Dabei wird das Virus dadurch blockiert, dass einige seiner Gene „zum Schweigen gebracht werden“ und deshalb nicht mehr funktionieren. Das heißt, die genetische Information von der DNA kann sich nicht mehr auf die mRNA übertragen, so dass die betreffenden Gene nicht abgelesen werden können (s. Chorea Huntington).
Das Virus ist damit funktionsunfähig.
Interview mit Prof. Theo Dingermann, Seniorprofessor für Pharmazeutische Biologie an der Universität Frankfurt. Seine Fachgebiete sind Biochemie und Molekularbiologie.
Zelltherapien, Gentherapien, Gen-Silencing: Herr Prof. Dingermann, wohin entwickelt sich die Arzneimittelforschung?
Prof. Dingermann: Schauen wir zunächst in die Schatzkiste unserer bisherigen Arzneimittel: Darunter findet sich kaum ein Medikament, das eine Krankheit heilen kann. Das heißt, die klassischen, synthetisch hergestellten Arzneimittel dienen im Großen und Ganzen dazu, Krankheiten zu managen. Denken Sie zum Beispiel an Medikamente gegen Bluthochdruck, Rheuma oder Diabetes. Es handelt sich dabei um Behandlungen, die so ausgelegt sind, dass die Patientinnen und Patienten zwar mit der Krankheit zurechtkommen und damit auch oft alt werden, aber im Grunde genommen halten die Medikamente nur die Symptome in Schach. Im Unterschied zu dieser, das Krankheitsgeschehen kontrollierenden Medizin, haben wir es bei den neuartigen Therapien mit einer reparierenden Medizin zu tun: Mutierte Gene werden ausgetauscht oder verändert. Körpereigene Immunzellen des Patienten so ausgestattet, dass sie zum Beispiel Krebszellen erkennen und bekämpfen können. Oder man repariert Knorpeldefekte durch knorpelbildende Zellen, die man dem Patienten entnommen hat. Das ist doch fantastisch! Damit hat eine neue Ära begonnen.
Diese neuartigen Therapien bestehen aus Zellen, Genen oder Gewebe – im Unterschied zu den chemisch hergestellten klassischen Arzneimitteln. Muss diesem Unterschied nicht auch das Zulassungsverfahren Rechnung tragen?
Prof. Dingermann: Bei den neuartigen Therapien – in Fachkreisen auch Advanced Therapy Medicinal Products, kurz ATMP genannt – handelt es sich tatsächlich um komplexes biologisches Material. Es sind lebende Medikamente! Das erfordert eine etwas andere Art der Zulassung. Ein synthetisch hergestelltes Molekül kann in beliebigen Mengen reproduziert werden, die Moleküle werden alle gleich sein. Also eine Aspirin-Tablette ist absolut identisch mit einer anderen. Wenn Forscher aber Knorpel anzüchten oder menschliche Zellen aufrüsten, dann sind die Produkte nicht ganz gleich – allein deshalb, weil sie von unterschiedlichen Menschen stammen. Man muss also mit einer gewissen Heterogenität umgehen. Deshalb hat sich in den letzten Jahren eine neue Philosophie herauskristallisiert: Basis der Zulassung ist bei biotechnologischen Produkten nicht nur das Produkt, sondern auch der Prozess der Herstellung. Der muss extrem konstant sein und unterliegt strikten Regularien.
Aber nichtsdestotrotz müssen die Therapien doch auf Wirksamkeit und Sicherheit für die Patientinnen und Patienten geprüft werden?!
Prof. Dingermann: Oft gibt es ja nur sehr wenige Patienten. Bei seltenen Erkrankungen sind es unter Umständen nur 50 weltweit. Dann muss sich die Forschung damit begnügen, nur an einer Handvoll Patienten zu testen, ob das Medikament bzw. das Produkt den gewünschten Effekt hat. Trotz solch kleiner Teilnehmerzahlen wird bei positivem Effekt eine Zulassung erteilt, um den oft schwerkranken Patientinnen und Patienten einen schnellen Zugang zur Therapie zu ermöglichen. Oft steht ihnen ja auch gar keine Behandlungsalternative zur Verfügung. Allerdings sind diese bedingten Zulassungen auch mit Auflagen verbunden, um die Risiken abzufangen. Die pharmazeutischen Unternehmen sind verpflichtet, in der Anwendung die Therapie weiterhin zu testen, und zwar unter Studienbedingungen. Das Verhältnis von Nutzen und Risiko wird damit nach der Zulassung regelmäßig überprüft.
Arzneimittel für immer weniger Patienten: Wird die Arzneimitteltherapie immer individueller?
Prof. Dingermann: Ja, das wird sie. Stichwort Personalisierte Medizin: Mit moderner Diagnostik können genetische, molekulare oder zelluläre Besonderheiten eines Patienten erfasst werden, um schon vor einer Therapie zu prüfen, ob bei diesem Patienten die Therapie überhaupt wirksam ist, oder ob er sie verträgt. Doch der Begriff „Personalisierte Medizin“ ist irreführend. Eigentlich müsste es Stratifizierte Medizin heißen. Stratifizierung bedeutet, dass man die Patientinnen und Patienten in Untergruppen einteilt: Bei welchen Betroffenen schlägt die Therapie an, bei welchen nicht, und wer muss mit gravierenden Nebenwirkungen rechnen? Tendenziell wird eine Therapie zukünftig immer im „Tandem“ mit einem solchen Vortest von statten gehen. Davon zu unterscheiden sind übrigens wirklich „persönliche Medikamente“ wie beispielsweise die CAR-T-Zell-Therapie. Die passt tatsächlich nur für einen einzelnen Patienten, weil sie aus seinen eigenen Zellen besteht.
Treiben Personalisierte Medizin und hoch individualisierte Medikamente nicht die Kosten im Gesundheitswesen in die Höhe?
Prof. Dingermann: Wenn man das gesamte Versorgungssystem in den Blick nimmt, sind neuartige Therapien nur auf den ersten Blick teuer. Systemisch betrachtet sind, sie oft effizienter und damit kostengünstiger. So kostet das bisher teuerste Medikament der Welt, Zolgensma, zwar zwei Millionen Euro – dafür sind die Kinder aber im besten Fall nach einer Spritze geheilt und müssen nicht über viele Jahre behandelt werden. Oder nehmen Sie das Beispiel der neuen Medikamente gegen Hepatitis C. Da wundern sich viele, warum eine Packung mehrere Tausend Euro kostet. Doch bei den allermeisten Patientinnen und Patienten sind nach drei Monaten Therapie keine Viren mehr im Blut nachweisbar, so dass sie nicht mehr lebenslang Medikamente einnehmen oder gar eine Lebertransplantation über sich ergehen lassen müssen.
Den Vorwurf, dass die pharmazeutischen Unternehmen eine Hochpreispolitik betreiben, finden Sie also unberechtigt?
Prof. Dingermann: Solche Vorwürfe sind zu kurz gedacht. Zum einen schultern die pharmazeutischen Unternehmen die teilweise immensen Forschungskosten, und das oft für nur wenige Patienten. Zum anderen gehen den Unternehmen mit heilenden Medikamenten die Einnahmen einer Dauertherapie verloren. Deshalb müssen die Hersteller ihre Preise auch anders kalkulieren. Dazu kommt, dass sie die absurd herunterregulierten Preise für Alt-Arzneimittel wettmachen müssen. Eine Aspirin-Tablette ist billiger als eine Zwiebel, das ist ein Unding! Wenn pharmazeutische Hersteller nicht zumindest im ersten Jahr nach der Zulassung für Innovationen hohe Preise verlangen könnten, gäbe es auch keine neuen Medikamente mehr für Patienten. Das ist eben der Deal.
Inwiefern bringt die Corona-Pandemie Schwung in die Arzneimittelforschung?
Prof. Dingermann: Bisher haben die Behörden oft wochenlang über den Unterlagen gesessen, und erst nach einem Bescheid wurde die nächste Phase eingeleitet. Jetzt spart man unglaublich viel Zeit, indem bestimmte Schritte parallel ablaufen und nicht nacheinander. Während eine Studienphase noch läuft, stimmt man sich schon über die nächste Phase ab. Zudem werden beispielsweise schon in frühen Studienphasen mehr Teilnehmerinnen und Teilnehmer eingeschlossen oder in einer Studie gleich mehrere neue Arzneimittel-Kandidaten gleichzeitig getestet. Sicherlich lernen die Zulassungsbehörden dadurch massiv dazu. Noch relevanter ist aber der Schub, den neue Technologien erfahren, die es bisher noch nicht auf dem Markt gibt. Ich denke da vor allem an die RNA-Impfung. Einen solchen Impfstoff in großen Mengen zu produzieren, ist eine Sache von Wochen statt bisher Monaten und zudem vergleichsweise günstig. Und sollte das Coronavirus mutieren, hätten wir auch dann innerhalb von wenigen Wochen einen neuen Impfstoff auf dem Markt.

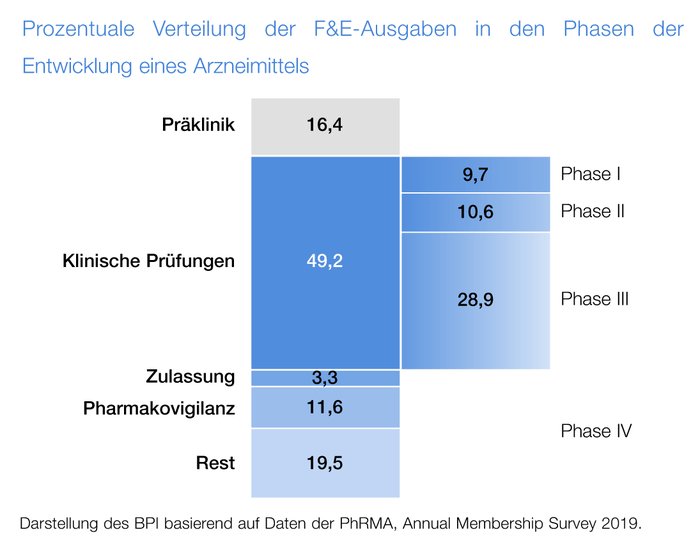
- die Ausgaben der Gesetzlichen Krankenversicherung für Arzneimittel – gemessen als Anteil am Bruttoinlandsprodukt – über viele Jahre stabil geblieben sind? Der Anteil der Arzneimittelausgaben am BIP liegt bei rund 1,16 Prozent.
- Deutschland auf Platz 2 liegt, was klinische Forschung angeht, hinter den USA? Innerhalb der EU ist Deutschland auf Platz 1.
- von 10.000 Molekülen, die zu Beginn einer Medikamentenentwicklung in Frage kommen, es gerade mal nur eine Substanz schafft, nach etwa acht bis zwölf Jahren den Zulassungsprozess erfolgreich zu absolvieren?
- im Bereich Lungenkrebs zwischen 1998 und 2014 zehn neue Medikamente eingeführt wurden, im gleichen Zeitraum aber 167 Studienprogramme abgebrochen werden mussten?
- dass die Krebssterblichkeit seit 1990 dank neuer Krebsmedikamente um 25 Prozent abgenommen hat?
- dass bei Leukämiepatientinnen und -patienten, bei denen alle Therapien versagt haben, nach der CAR-T-Zell-Therapie der Tumor nicht mehr nachweisbar war?
Krebsgesellschaft/Onko-Internetportal: „Zielgerichtete Krebstherapien/Die Molekularbiologische Therapie bei Krebs“
https://www.krebsgesellschaft.de/onko-internetportal/basis-informationen-krebs/therapieformen/molekularbiologische-therapie.html
Deutsches Krebsforschungszentrum/Krebsinformationsdienst: „Immuntherapie gegen Krebs: Die körpereigene Abwehr nutzen“, Informationsblatt
https://www.krebsinformationsdienst.de/service/iblatt/iblatt-immuntherapie.pdf
Deutsches Krebsforschungszentrum/Krebsinformationsdienst: „Neue Krebstherapien: Zielgerichtete Therapie, Immuntherapie, Virotherapie“, Informationsblatt
https://www.krebsinformationsdienst.de/service/iblatt/iblatt-neue-krebstherapien-ueberblick.pdf
Deutsches Krebsforschungszentrum/Krebsinformationsdienst: „Zielgerichtete Krebstherapie: Das Tumorwachstum punktgenau hemmen“
https://www.krebsinformationsdienst.de/behandlung/gezielte-krebstherapie.php
Deutsches Krebsforschungszentrum/Krebsinformationsdienst: „Update CAR-T-Zell-Therapie“
https://www.krebsinformationsdienst.de/fachkreise/nachrichten/2019/fk19-car-t-zell-therapie-jahres-update2019.php
Pharmazeutische Zeitung: „Schwerpunkt lag wieder auf Krebs“, Artikel von Daniela Hüttemann vom 16.12.2019
https://www.pharmazeutische-zeitung.de/schwerpunkt-lag-wieder-auf-krebs/
Pharmazeutische Zeitung: „ATMP: Herausforderungen, Chancen, Risiken“, Artikel von Nicole Schuster vom 26.6.2019
https://www.pharmazeutische-zeitung.de/herausforderungen-chancen-und-risiken/
Technische Universität München: „Verbesserung der Immuntherapie bei Krebs“, Pressemitteilung vom 29.4.2020
https://www.tum.de/nc/die-tum/aktuelles/pressemitteilungen/details/35998/
vfa, Verband Forschender Arzneimittelhersteller e.V. (Hrsg.): „Wie aus Wissen medizinischer Fortschritt wird. Neue Krebsmedikamente“ Broschüre
https://www.vfa.de/download/wie-aus-wissen-medizinischer-fortschritt-wird.pdf
Gene ersetzen, reparieren oder stilllegen: Seltene Erkrankungen
Bundesverband der Pharmazeutischen Industrie (BPI) e.V.: „Orphan Drugs“
https://www.bpi.de/de/alle-themen/orphan-drugs
Der Tagesspiegel: „Glücksfall Gentherapie: Die Lotterie des Lebens“, Artikel von Thomas Trappe vom 4.2.2020
Der Spiegel: „Was hinter der Verlosung eines Millionen-Medikaments steckt“, Artikel von Stefan Schultz und Nina Weber vom 2.2.2020
https://www.spiegel.de/wirtschaft/soziales/zolgensma-novartis-verlost-teuerstes-medikament-der-welt-an-kinder-a-44556bb4-dece-448e-8917-4d397910f483
Deutsche Gesellschaft für Neurologie: „Phase 1-2a-Studie zur kausalen Therapie der Chorea Huntington gibt Anlass zur Hoffnung“, Pressemitteilung vom 7. Mai 2019
https://www.dgn.org/presse/pressemitteilungen/60-pressemitteilung-2019/3775-phase-1-2a-studie-zur-kausalen-therapie-der-chorea-huntington-gibt-anlass-zur-hoffnung
Handelsblatt: „Zwei-Millionen-Gentherapie von Novartis kann bald zugelassen werden“, Artikel von Maike Telgheder vom 27.3.2020
https://www.handelsblatt.com/unternehmen/industrie/zolgensma-zwei-millionen-gentherapie-von-novartis-kann-bald-zugelassen-werden/25690516.html?ticket=ST-7097524-HlDbgXq4vny6yRSTf3NL-ap5
vfa, Verband Forschender Arzneimittelhersteller e.V. (Hrsg.): „Forschung für die Waisen des Gesundheitssystems. Medikamente für Menschen mit seltenen Erkrankungen“, Broschüre, Stand: Februar 2019
https://www.vfa.de/download/forschung-fuer-die-waisen-des-gesundheitssystems.pdf
vfa, Verband Forschender Arzneimittelhersteller e.V.: „Was der Orphan Drug-Status für ein Medikament bedeutet (und was nicht)“, Artikel vom 16.2.2018
https://www.vfa.de/de/wirtschaft-politik/artikel-wirtschaft-politik/was-der-orphan-drug-status-fuer-ein-medikament-bedeutet.html
Das Tal des Todes umschiffen: Wie schwierig ist Arzneimittelforschung?
Deutsche Krebshilfe: „Klinischen Studien“, eine Broschüre aus der Reihe „Die blauen Ratgeber“
https://www.krebshilfe.de/infomaterial/Blaue_Ratgeber/Klinische-Studien_BlaueRatgeber_DeutscheKrebshilfe.pdf
Deutsches Krebsforschungszentrum/Krebsinformationsdienst: „Klinische Studien: Was muss ich wissen?“, Informationsblatt
https://www.krebsinformationsdienst.de/service/iblatt/iblatt-klinischestudien.pdf
Deutsches Zentrum für Infektionsforschung (DZIF): „Entwicklung von Medikamenten: Der lange Weg zum neuen Medikament“
https://www.dzif.de/de/entwicklung-von-medikamenten
Helmholtz (Hermann von Helmholtz-Gemeinschaft Deutscher Forschungszentren e.V.): „Neue Medikamente: Der lange Weg zum Patienten“
https://www.helmholtz.de/gesundheit/der-lange-weg-zum-patienten/
Preisbildung – Komplexe Regularien – Innovationshürden
Arzneimittelkommission der deutschen Ärzteschaft: „Frühe Nutzenbewertung nach AMNOG und Auswirkungen auf die Vertragsärzte“, Artikel von Britta Bickel
https://www.akdae.de/Arzneimitteltherapie/AVP/Artikel/201601/043h/index.php
arzt-wirtschaft.de: „Regulierungsdichte und Kosten weiterhin hoch: Pharmaindustrie unter Druck“, 10.1.2020
https://www.arzt-wirtschaft.de/regulierungsdichte-und-kosten-weiterhin-hoch-pharmaindustrie-unter-druck/
Bundesverband der Pharmazeutischen Industrie (BPI) e.V. (Hrsg.): „Pharma-Daten 2019“, Broschüre
https://www.bpi.de/fileadmin/user_upload/Downloads/Publikationen/Pharma-Daten/Pharma-Daten_2019_DE.pdf
Bundesverband der Pharmazeutischen Industrie (BPI) e.V.: „Die pharmazeutische Industrie – ein industrieller Kern der Gesundheitswirtschaft“, Positionspapier von 2015
https://www.bpi.de/fileadmin/user_upload/Downloads/Publikationen/Positionen/BPI-Positionspapier-PharmaIndustrie-2015.PDF
Corona – Beschleunigte Forschung
Deutsche Herzstiftung (Hrsg.): „In den Zeiten von Corona. Fragen und Antworten“, in: Herz heute 2/2020.
Der Tagesspiegel: „Der lange Weg zum Impfstoff“, Artikel von Sascha Karbert vom 25.5.2020
Der Tagesspiegel: „Ein riskantes Rennen“, Artikel von Richard Friebe vom 25.5.2020
Der Tagesspiegel: „Mit allen Mitteln“, Artikel von Edda Grabar vom 9.4.2020
Die Bundesregierung: „Bundesregierung beteiligt sich mit 300 Millionen Euro an Curevac“, Pressemitteilung vom 15.6.2020
https://www.bmwi.de/Redaktion/DE/Pressemitteilungen/2020/20200615-bundesregierung-beteiligt-sich-mit-300-millionen-euro-an-curevac.html
Julius-Maximilians-Universität Würzburg: „Intensive Forschung zu SARS-CoV-2“, Pressemitteilung vom 21.4.2020
https://www.med.uni-wuerzburg.de/aktuelles/meldungen/single/news/intensive-forschung-zu-sars-cov-2/
Max-Planck-Gesellschaft: „Die Zacken in der Viruskrone“, Artikel vom 6.4.2020
https://www.mpg.de/14652142/corona-spike-protein
Redaktionsnetzwerk Deutschland: „Von Vektor- bis DNA-Impfstoff: Wie die Forschung das Coronavirus bekämpfen will“, Artikel von Laura Beigel, 24.5.2020
https://www.rnd.de/gesundheit/von-vektor-bis-dna-impfstoff-wie-die-forschung-das-coronavirus-bekampfen-will-LSDK3CECMJE4FINQJF5QVSKK34.html
Spektrum der Wissenschaft: „Vielleicht wird es keinen Corona-Impfstoff geben“, Artikel von Annika Röcker vom 8.6.2020
https://www.spektrum.de/news/vielleicht-wird-es-keinen-corona-impfstoff-geben/1740288
vfa, Verband Forschender Arzneimittelhersteller e.V.: „Impfstoffe zum Schutz vor Covid-19, der neuen Coronavirus-Infektion“
https://www.vfa.de/de/arzneimittel-forschung/woran-wir-forschen/impfstoffe-zum-schutz-vor-coronavirus-2019-ncov
vfa, Verband Forschender Arzneimittelhersteller e.V.: „Therapeutische Medikamente gegen die Coronavirusinfektion Covid-19“
https://www.vfa.de/de/arzneimittel-forschung/woran-wir-forschen/therapeutische-medikamente-gegen-die-coronavirusinfektion-covid-19